Reports: AC3
48546-AC3 Probing the Articulation Between Electron Transfer and Bond Cleavage Using Early Metal Complexes of Redox-Active Phenoxides
The chemical heart of a fuel cell is a catalyst that can mediate between the worlds of single-electron outer-sphere transfer relevant to passing current, and that of multi-electron inner-sphere oxidations and reductions involved in making and breaking the chemical bonds of fuel and oxidant. This research explores a new way of mediating between these two worlds, using a metal complex where the organic ligand undergoes one-electron redox chemistry, and the metal atom holds the substrate in physical and electronic contact with the ligand and facilitates its changes in bonding.
New diarylaminoaryloxide ligands and their complexes
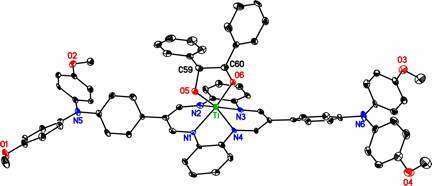
Our initial target involved tetradentate, dianionic N2O2 ligands of the salan type. N,N'-bis(5-bromo-2-hydroxybenzyl)-N,N'-dimethylethylenediamine is readily prepared from commercially available 5-bromosalicylaldehyde. Protection of the phenols as tert-butyldimethylsilyl ethers, followed by Hartwig-Buchwald amination using bis(4-methoxyphenyl)amine and desilylation, affords the corresponding salan ligand with two redox-active di(4-methoxyphenyl)aminoaryloxides.
This
ligand binds titanium well, and we have prepared and characterized a number of
complexes with potentially oxidizable ancillary ligands such as secondary
alkoxides (isopropoxide), 1,2-diols (1,1,2-triphenylethanediolate), and alpha-hydroxyacids
(2-hydroxyisobutyrate). In general
outer-sphere oxidation results in long-lived green radical cations, but only in
the case of 1,1,2-triphenylethanediolate is substrate oxidation (to
benzaldehyde and benzophenone) observed.
Unfortunately, we have been unable to synthesize any other 1,2-diolate
complexes, hampering attempts to assess the generality of the reaction or to
study structure-activity relationships.
The ligand also appears to be rather flexible, adopting symmetrical cis-alpha or unsymmetrical cis-beta arrangements depending on the nature of the
ancillary groups.
In
order to reduce the structural flexibility of the framework, we have begun to
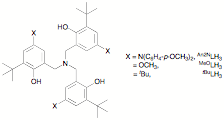
examine complexes of tripodal aminetriphenol ligands LH3. These ligands have the added advantages
of conferring excellent water-stability on their early metal complexes, and of
allowing an open coordination site in complexes LTiX. Both these features are likely to be important in allowing
catalytic applications, particularly under ambient (i.e., wet) conditions. We have made a
di-(4-methoxyphenyl)amino-substituted tripodal ligand An2NLH3
using chemistry similar to that used for the salan ligand, and have compared
its properties with the methoxy-substituted MeOLH3 that
we recently prepared and the well-known tert-butyl substituted tBuLH3.
Cyclic voltammetry studies of the six-coordinate LTi(acac) complexes illuminate the effect of the diarylamino substituent on the redox properties of the metal complexes. While tBuLTi(acac) shows only one reversible oxidation, MeOLTi(acac) shows two reversible oxidations separated by about 350 mV. In addition to oxidizing at a substantially lower potential, An2NLTi(acac) shows reversible oxidations for each of the three arms, separated by about 150 mV from each other. (In fact, since each of the arms also shows a second one-electron oxidation at about 700 mV higher potential than the first, An2NLTi(acac) can effectively store up to 6 oxidizing equivalents.) This validates a number of features of the diarylaminophenoxide ligand design. In order to minimize overpotentials, an ideal electrocatalyst for a multielectron reaction should gain or lose all its electrons at about the same potential. By moving the locus of oxidation farther from the metal, An2NLTi(acac) reduces the difference in potential between successive oxidations. At the same time, the retention of a modest electronic coupling between the aryloxide groups suggests that there is enough electronic communication between the radical centers and the titanium to allow ligand oxidation to affect the chemistry of other groups coordinated to the metal center. Our attention is now focusing on stoichiometric or catalytic chemistry of unsaturated An2NLTi complexes. Preliminary results indicate that An2NLTiCl does undergo slow, but relatively selective, ligand oxidation in the presence of air. In contrast, neither the saturated An2NLTi(acac), nor the unsaturated but less easily oxidized MeOLTiCl, shows any reactivity towards O2.
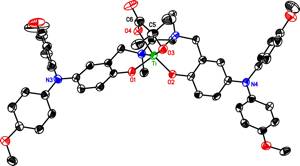
We are also exploring the use of these tripodal ligands to support oxometal fragments where the oxo group can bind a proton with concomitant one- or two-electron redox changes at the ligand to allow proton-coupled electron transfer reactions. Such reactions typically involve redox changes at metal centers coupled with proton transfer to ligands; our use of ligands to carry out both changes is unusual (though precedented, for example, by oxometal complexes of porphyrin radical cations that likely carry out oxidations in cytochromes P450). So far, we have found that LMo(O)2– and LW(O)2– salts can be prepared directly from molybdate and tungstate under mild conditions, which emphasizes their hydrolytic stability. Compounds with the tert-butyl and methoxy-substituted ligands may be reversibly oxidized by one (but only one) electron, and all can be protonated reversibly by mild acids (e.g., saccharin). Combining the pKa and Eo data indicates that the neutral radicals LMo(O)2 and LW(O)2 would be mild hydrogen atom acceptors, but so far the neutral species have proven too unstable to explore their chemistry. Hopefully the An2NL derivatives of these metals (currently being synthesized) will be more stable in the oxidized form, and may allow us to explore the more interesting possibility of two-electron PCET.
Redox reactivity of novel titanium tetraazaannulenes
As
a complementary ligand scaffold, we have been exploring
6,13-disubstituted-tetraazaannulenes (depicted below). The analogous
5,7,12,14-tetramethyl-substituted compounds had been reported to have excellent
water stability and interesting reactivity, but only one brief report of the
less substituted TAA titanium complexes had appeared. We find that the ligands are easily synthesized and that the
metal complexes are indeed water-stable, though the oxometal species tend to
precipitate irreversibly on standing, possibly through Ti=O--Ti=O polymer
formation.
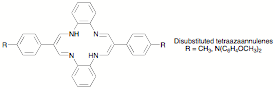
In this system a wide variety of 1,2-diolate as well as a-hydroxycarboxylate complexes can be prepared. Outer-sphere oxidation generally leads to high yields of carbon-carbon bond cleavage products, but rates vary substantially, with the tolyl derivatives reacting rapidly and the dianisylamino compounds more slowly. The relatively slow reaction rates of the latter compounds will be explored in more detail to study the kinetics of bond cleavage and to explore structure-activity relationships. Of particular interest is whether the slower rates are due to lower redox potentials (decreased driving force), or whether moving the locus of oxidation farther away from the metal makes the reaction kinetically inefficient.