Reports: G6
46735-G6 Theoretical Investigation of the Two-state Model for the Excess Electron in Saturated Hydrocarbon Liquids Based on Path Integral Simulation
During the last funding period, the principal investigator (PI) made significant progress in the path integral centroid molecular dynamics (CMD) simulation of an excess electron in supercritical helium, and has been making steady advances in the development of path integral simulation codes for an electron in hydrocarbon liquids.
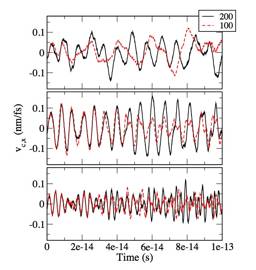
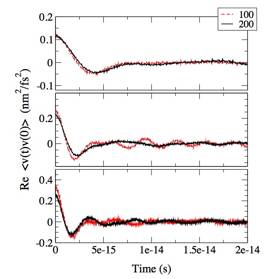
Figure 1 shows path integral centroid velocities (x-components) of an electron in helium solvents with different values of the reduced density (the number of helium atoms per cube of Lennard Jones radius of helium), 03, 0.5, and 0.7. Time evolution in the CMD method requires averaging of centroid forces at each time over sufficient number of paths for each constrained value of centroid evolving in time. Two trajectories with different sampling numbers, 100 and 200, are shown in Fig. 1. While the trajectories diverge in the long run, within the time scales of velocity correlation, the difference between the two trajectories is relatively small. Figure 2 shows quantum velocity correlation functions calculated from single CMD trajectories shown in Fig. 1, and confirms that the difference between the two numbers of samplings is relatively minor. Thus, a reasonable convergence is expected with number of samplings less than 1,000, which is feasible.
Additional samplings of centroid trajectories need to be made over different initial conditions. It is expected that about 100 of such initial conditions are sufficient for producing reliable CMD data. This will be the first CMD simulation ever for the problem of an excess electron. Recently, a ring polymer molecular dynamics (RPMD) simulation was performed for the same system by another researcher. Comparison of the two results will provide great insights into the relative performance of the two methods for this challenging quantum dynamical problem. All the simulation methodologies and experiences gained from this system can be extended for methane and larger hydrocarbon liquids.
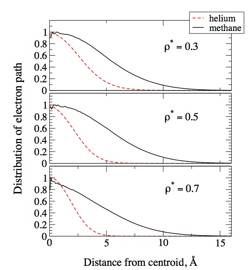
Path integral molecular dynamics simulation code for an excess electron in methane has been developed and is being tested. Large enough discretization of electron path, 1,400, and number of solvents, 2,048, were chosen. Figure 3 shows distributions of electron paths relative to the centroid, which indicate the extent of delocalization, for three values of reduced densities. Results for helium are shown as well. As expected, the distributions of electron paths are broader and insensitive to the concentration in methane when compared with those in helium. This indicates that electron in methane behaves almost like a free and thermalized particle. Whether the CMD method can provide reasonable description of the dynamics of such electron is an interesting issue. Extension of developed path integral code for CMD simulation is straightforward and can be complete soon. Test and comparison with experimental data will provide a good assessment of the reliability of the CMD method for delocalized electrons in weakly perturbing solvents.
In collaboration with a postdoctoral researcher, model potentials between electron and propane, butane, and pentane are currently being developed. A series of path integral simulations will follow the completion of these model potentials. These simulations are aimed at understanding how the structure, configuration, and dynamics of solvents correlate with the energetic and dynamic features of the electron. CMD simulation will be performed for these hydrocarbon liquids and will be analyzed in accordance with the assessments made for two simpler extreme environments, helium and methane. All of these simulation results will be essential in understanding the mechanistic details of the dynamics of electron and in determining whether the simple two-state picture of dynamics is indeed valid.
In parallel with the path integral simulation projects described above, substantial progress has been made in theory development and computational modeling of electronic resonance energy transfer processes. While this subject is not directly related to the dynamics of excess electron in hydrocarbon liquids, it has fundamental importance in the utilization of unsaturated hydrocarbon molecules for solar energy conversion. A paper on this project, which is a continuation of the work published in the last funding period, was recently accepted for publication in the Journal of Chemical Physics. Application of this theory to the modeling of pump-probe anisotropy spectroscopy of dithia-anthracenophane system is currently in progress. This will help to elucidate the role of quantum coherence in electronic resonance energy transfer processes.
The present research project played a critical role in generating other major research funds. This year, the PI was awarded a CAREER grant from the National Science Foundation (NSF) on a project titled "Synergistic theory development and computational modeling of soft optoelectronic molecules." Another related project also being funded by the Department of Energy (DOE) is "Computational modeling and theory development of energy and charge flow dynamics in photosynthetic units and conjugated polymer systems." The objective of this proposal is quantitative modeling of energy and charge flow dynamics based on the combination of Quantum Master equation (QME) theories and simulations. Most recently, the PI submitted a proposal for the Early Career program of DOE with a title, "Multiscale quantum dynamics simulation of electron and hole migration in soft disordered environments." This project includes various types of computational and simulation methods for the description and understanding of charge flow dynamics across different length and time scales in systems like bulk heterojunction plastic solar energy conversion devices. Path integral simulation plays a key role in this future project, and the experience gained through the present funding has been essential in successful completion of the DOE proposal.
|
|
|
|||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||