Reports: UNI3
49510-UNI3 Chirality Recognition by Macrocyclic and Multi-Functional Metal Complexes
Summary:
We have initiated syntheses of various macrocyclic ligands containing the trans-1,2-diaminocyclohexane (DACH) moiety both as a source of chirality, and a metal chelating unit. Two such compounds have currently been isolated ((R)-1 and (R)-2), while similar targets are being pursued. We are also optimizing the final macrocyclization steps in the syntheses of (R)-1 and (R)-2, and have explored the binding of the obtained compounds to Zn(II) and Cu(II) ions. Both ligands appear to to bind Cu(II) very weakly, but have high affinities for Zn(II). The binding of (R)-1 to Zn(II) in MeOH has been monitored by 1H NMR, and a crystal structure of the complex (R)-1:ZnCl2 has been determined. The free ligand 1 was also found to differentiate the enantiomers of mandelic acid (MA) by 1H NMR in d-CHCl3. When racemic MA was titrated with (R)-1, differentiation of MA enantiomers with baseline resolution of the PhCH(OH)CO2H signals was obtained, suggesting that macrocycles such as (R)-1 may be generally useful as chiral shift reagents for a-chiral carboxylic acids.
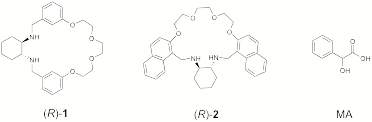
Organic Synthesis:
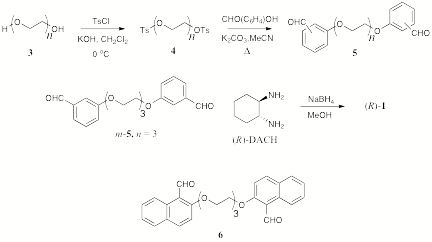
The synthetic approach to DACH-based macrocycles is illustrated in Scheme 1. An oligoethylene glycol 3 (n = 3 or 4) is di-tosylated to give 4, and subsequently converted to a dialdehyde such as 5 by nucleophilic substitution of the tosylates by phenoxide substituents. The macrocyclization is then accomplished through a reductive amination reaction with DACH, in which the di(imine) precursor is generated under pseudo high dilution conditions. The required dialdehyde subunits (o, m, and p-5, n = 3, 4, and 6) are readily synthesized in high yields (75-85% over two steps). The macrocyclization reaction appears to work well; as in all cases studied, the 1+1 macrocycle appears by TLC to be the dominant product. However, the separation of this species from larger macrocycles and oligomers by column chromatography has been problematic, as very little (typically 10-20 %) of the material initially loaded onto the column is recovered. The Rf values the DACH-based macrocycles are very low, even when very polar eluents such as MeOH are used. SiO2 is found to be a preferable to Al2O3 as a stationary phase, but attempts at stationary phase deactivation by treatment with water and tertiary amines has yet to produce reproducible separation with high yields. Attempts to purify the macrocycles by Kugelrohr distillation have failed.
Scheme 1.
Metal Complexation: Initial efforts focused on the chelation of Cu(II) by the purified macrocycles. This metal ion is of particular interest because of its kinetic lability, tendency to form square planar complexes, and electronic absorption band in the visible region. Attempts to quantify the binding of Cu(OTf)2 by (R)-1 by spectrophotometric titration lead to a linear increase in the intensity of an absorption band at 588 nm with increasing [(R)-1] well past 1 equivalent in both 1:1 MeOH:H2O (10 mM HEPES buffer, pH = 7.2) and pure MeOH, suggesting very weak binding. A similar result in MeOH was obtained for (R)-2. On the other hand, (R)-1 cleanly forms a 1:1 complex with ZnCl2 in d4-MeOH, as evidenced by 1H NMR titration. The emergence of distinct signals for the metal complex with the simultaneous disappearance of those for the free ligand demonstrates that Zn(II) exchange is slow in the 1H NMR timescale (Figure 1A).
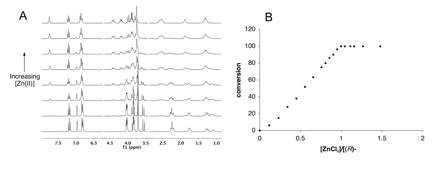
Figure 1. A. Changes in the 300 MHz 1H NMR spectrum of (R)-1 (6.6 mM in d4-MeOH) upon titration of 0-1.5 eq. ZnCl2. B. Percentage of (R)-1 existing as the Zn(II) complex as a function of ZnCl2: (R)-1 ratio as determined from the integrated signal intensities of the ligand and the complex.
Crystallization of the (R)-1:ZnCl2 complex from MeOH allowed for the solid-state structure to be determined by X-ray crystallography (in collaboration with Professor Joseph Tanski of Vassar College). Two very similar crystallographically independent complexes are present in the unit cell. Figure 2 shows an ORTEP representation of one of these. The structure validates the design principles enumerated in the grant proposal; namely the binding of a labile metal ion within a well defined chiral macrocyclic cavity. It is imagined that additional (guest) ligand may be accommodated by dissociation of the Zn-Cl bonds. The complex (R)-2:ZnCl2 was found to be very insoluble in MeOH, but soluble in chloroform. Attempts to obtain X-ray quality crystals of (R)-2:ZnCl2 are presently underway.
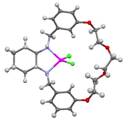
Figure 2. X-ray structure of the (R)-1:ZnCl2 complex. Thermal ellipsoids are scaled to 50% probability.
Enantioselective Binding of Mandelic Acid (MA): It was found that the addition of (R)-1 to a racemic sample of MA in d-CHCl3 differentiates the 1H NMR resonances of the MA enantiomers. The effect is most evident for the PhCH(OH)CO2H signal, which undergoes upfield shifting and splitting into two separate baseline-resolved peaks with the addition of ca. 0.1-0.4 equivalents of (R)-1. A maximal peak separation of > 0.06 ppm is achieved. Increasing [(R)-1] further causes additional upfield shifting of the signals (Figure 3A), with an eventual coalescence at [(R)-1]/[MA] > 0.5. The direction of the shift and the apparent 2:1 (MA: (R)-1 binding stoichiometry implies that MA is being deprotonated by the amine groups of (R)-1. Presumably, the resultant ammonium and carboxylate ions associate through Columbic forces and hydrogen bonding. This assumption is confirmed by the loss of chiral discrimination upon the addition of 10% d4-MeOH by volume.
Similar titration experiments using single enantiomers of MA show upfield shifting of a single PhCH(OH)CO2H resonance, with a greater response in the case of (R)-MA (Figure 3B). The binding isotherms show both inflection at about 0.5 equivalents of added (R)-1. Efforts to unambiguously determine binding stoichiometry by Job's method of continuous variation, and to extract binding constants by fitting the titration data presented in Figure 3B to a 2:1 binding model are underway.
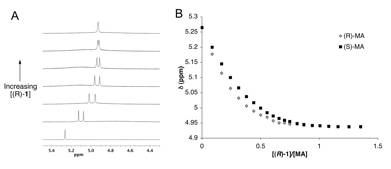
Figure 3. A. Part of the 300 MHz 1H NMR spectrum of rac-MA (7.1 mM) upon increasing [(R)-1] from 0 – 5.4 mM in d–CHCl3. B. Chemical shifts of enantiomerically pure MA samples (each 2.2 mM in d–CHCl3) upon titration with (R)-1.



