Reports: G1
47565-G1 Development of Carbon-Carbon Bond Activation for Organic Synthesis
Funding from the ACS-PRF has allowed us to develop a research program in catalytic C–C s-bond activation and functionalization. This chemistry is rare, with most examples prior to our work using strained C–C s-bonds. Our initial proposal was to develop activation of C–C bonds adjacent to ketones to explore non-traditional reactivity from this standard functional group. Our initially proposed research was aimed at activating simple ketones by using the principles of organocatalysis to form the corresponding ketimine and performing organometallic catalysis to functionalized the C–C bond via chelation-assisted bond activation (Scheme 1, left). Our initial work in this area proved somewhat difficult. After identifying that the initial ketimine formation step was a problem step, we made the early tactical decision to move examine feasibility of the organometallic catalysis portion. In this mode, the activation and functionalization takes place when a chelating heteroatom is embedded in the ketone substrate (7 to 8, via 9, Scheme 1, right). We have termed this net addition of a carbon and acyl substituent across an alkene "carboacylation"
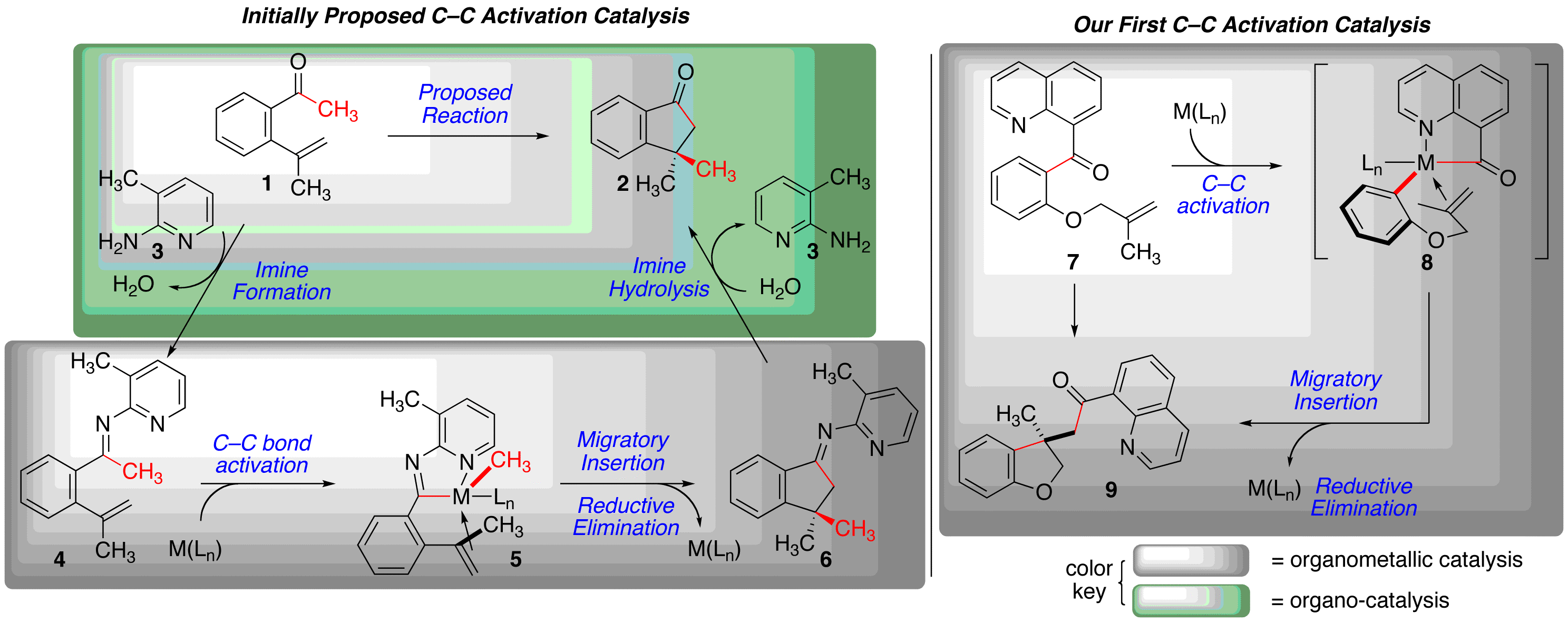
Scheme 1: Our initially proposed reaction, and our first successful C–C activation chemistry.
As the primary focus of our initial proposal was on C–C activation rather than imine-based catalysis, we decided to learn what we could from the 8-acyl quinoline system before re-investigating the original hybrid mechanism proposal. Graduate student Ashley Dreis made the initial discovery of 8-acyl quinoline promoted carboacylation (Scheme 1, right), finding that a suite of rhodium catalysts worked well with her initial test substrate (Scheme 2, left). After preparing a set of additional substrates, many of which are available in just a few (3–5) steps from salicylaldehyde, Ashley made a preliminary examination of the scope with respect to olefin substitution and linkage (heteroatom, aromatic, and tether) to the ketone (Scheme 2, right). All 1,1-disubsituted alkenes worked well, but monosubsituted phenolic ethers tended to undergo cleavage to the corresponding phenol under the reaction conditions, and provided only a low yield of the carboacylation product (15, Scheme 2).
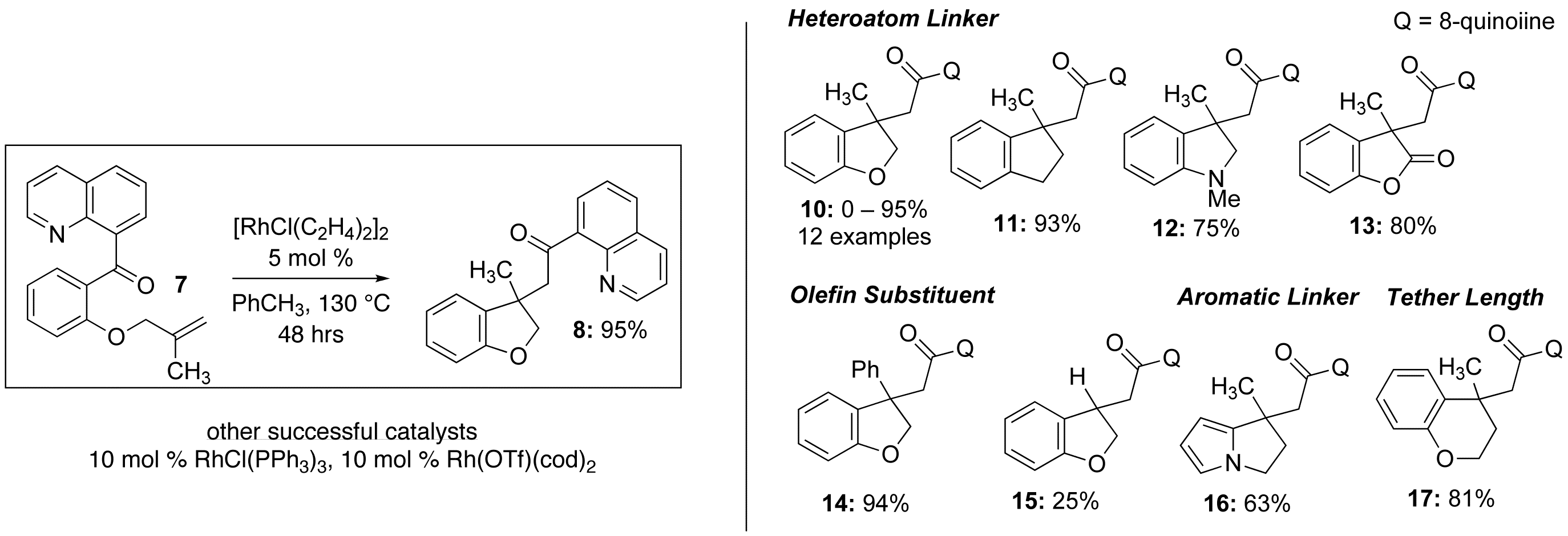
Scheme 2: Intramolecular carboacylation via quinoline-directed C–C activation.
Graduate students V. Jagan Reddy and Michael Wentzel, along with undergraduate Todd Hyster looked to expand this chemistry to intermolecular systems. Using the same catalyst and conditions that Ashley was successful with in the intramolecular carboacylation, Michael observed the product resulting from C–H activation ortho to the aryl ketone group and subsequent hydroarylation of norbornene in good yield (18 to 19, 79%). Fortunately, after further catalyst and condition screening, Michael was able to produce the desired compound (20) with no trace of 19 – a complete reversible of chemoselectivity! (Scheme 3) As a group, Jagan, Michael, Todd, and I were then able to further probe scope and limitations of the system. Our rudimentary understanding of the system is as follows: C–H activation is favored with coordinating catalyst counter-ions (Cl) and non-polar solvents (PhMe), while C–C activation is favored with less coordinating catalyst counter-ions (triflate) and more polar solvents (THF). The key relative rate between the two pathways is likely migratory insertion. Graduate student Ross Moran attempted similar intermolecular reactions of alkynes, but unfortunately, no positive results were obtained with either C–H or C–C activation reaction manifolds.
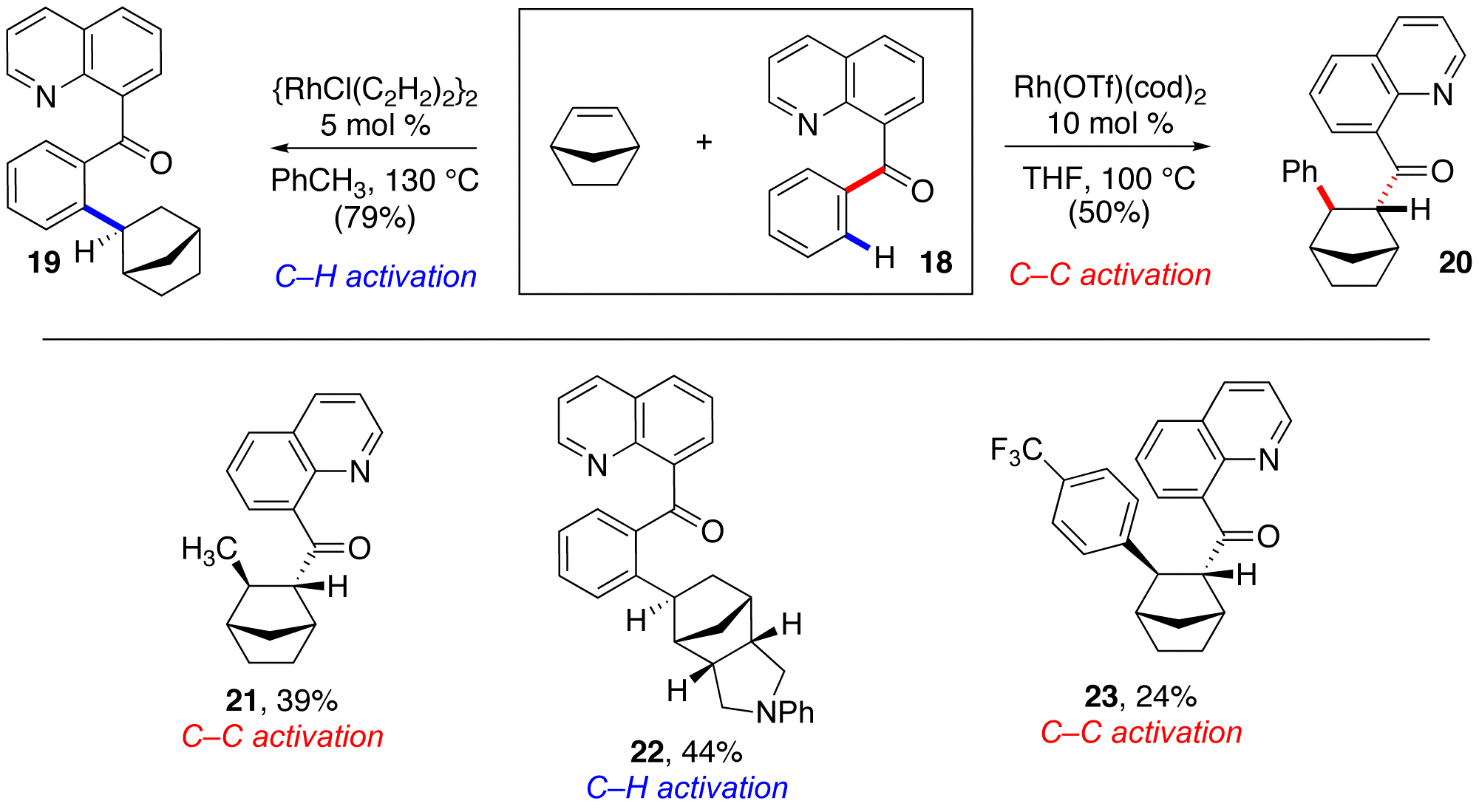
Scheme 3: Intramolecular carboacylation and competitive C–H activation.
Last summer, undergraduate student Michael Capp garnered some important preliminary data on intramolecular carboacylation of alkynes via C–C bond activation. We were initially concerned that salicylaldehyde-derived 8-acyl quinolines containing propargylic phenolic ethers would be prone to the same type of rhodium-promoted ether cleavage (to 26, Scheme 4) that plagued the formation of 15 in Ashley's work. After examining several reaction conditions, Michael Cap was able to identify a product resulting from alkyne carboacylation. Presumably, the first-formed carboacylation product (27) isomerized to the aromatic benzofuran under the reaction conditions. Although the isolated yields are low at this time, this is likely just due to purification difficulties; the ratio of product:phenol:starting material in the crude reaction mixture (2:3:0), with a relatively clean reaction indicates a promising lead. Michael Wentzel and Ashley Dreis are following up on these results, examining additional catalyst/condition optimization and other substrates (i.e. 28 & 29) that should be less prone to the side reactions observed by Michael Capp.
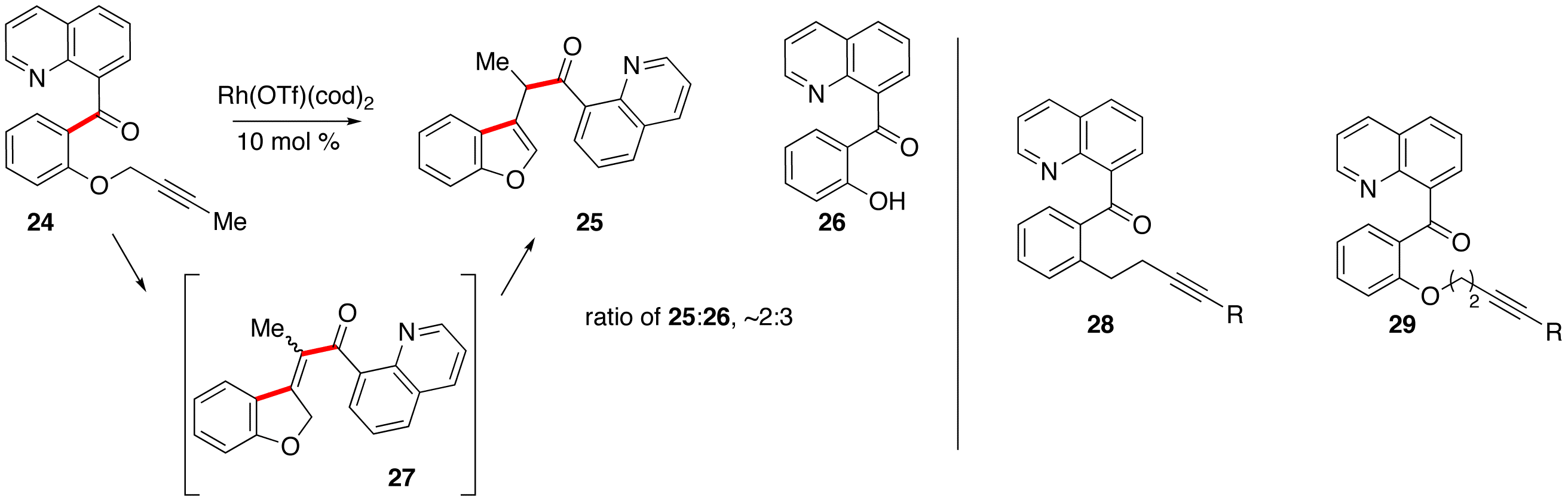
Scheme 4: Alkyne carboacylation
Graduate student T. Giang Hoang initially started to probe the potential for carboacylation of ketones via C–C activation. The outlined experiments were along the lines of Ashley's initial successes with alkenes (30 to 33 via 31). Giang typically saw low conversion in this attempted experiments, so he attempted a reaction with a stoiciometric amount of Wilkinson's catalyst. Surprisingly, we isolated a stable rhodium-phosphine complex, and tentatively assigned the structure as 32 based on spectroscopy. Even more surprisingly, the complex forms in 82% yield (by 1HNMR) at temperatures as low as –20 °C! We are uncertain about the exact coordination geometry at the rhodium center. Giang's results lead us to question whether the final proposed C–O reductive elimination was actually feasible in this system. Phrased another way, we wondered if catalytic quinoline-directed C–O activation of esters would be a possible (such as 33 to 32), and if so, would it allow an entry into "oxy-acylation" of alkenes. Giang undertook a study into this reaction manifold and has recently garnered some promising catalytic results, showing TON up to 8 for intramolecular oxyacylation. Giang is now looking into the scope and limitations of this reaction as well as examining chiral bidentate phosphine ligands for asymmetric induction at the newly formed stereocenter.
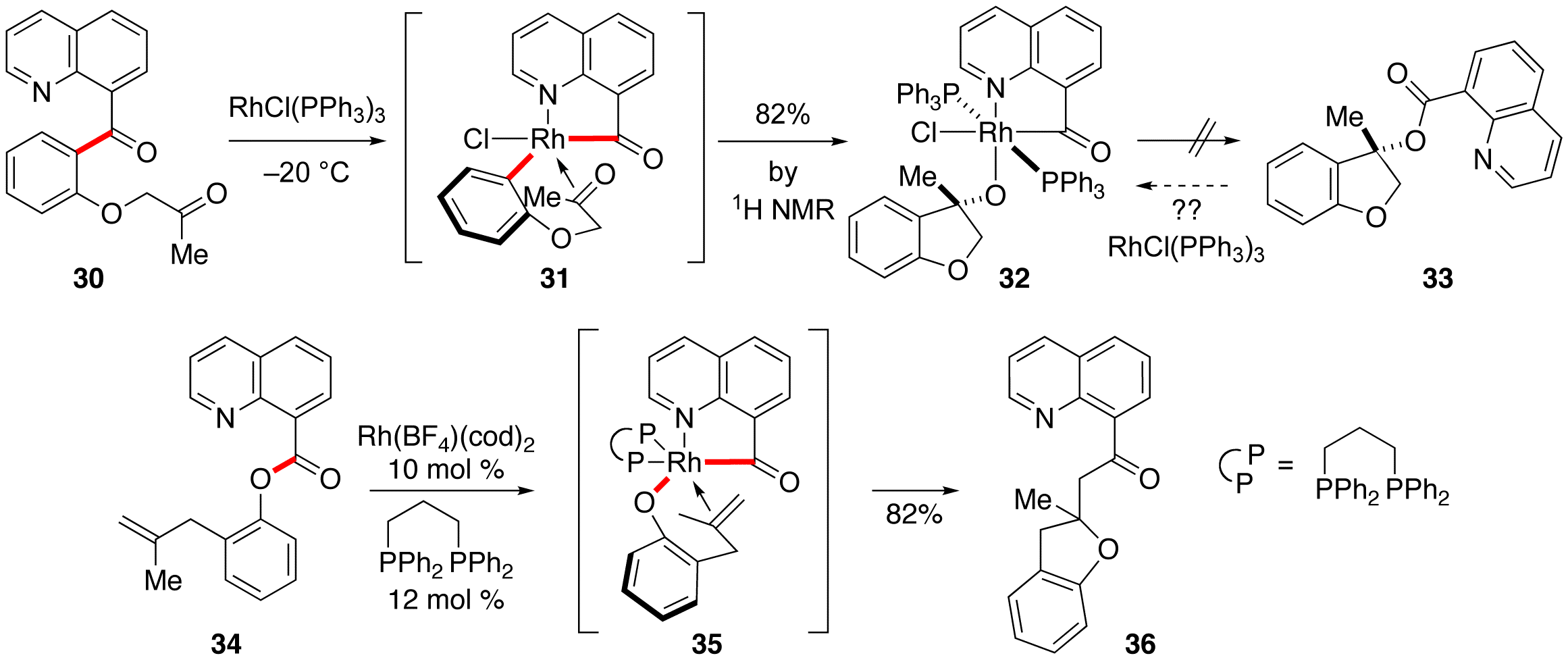
Scheme 5: Attempted Ketone Carboacylation and C–O Activation allows "Oxy-Acylation" of Alkenes
Although our initial hypothesis regarding the 2-amino-pyridine based co-catalysts (Scheme 1) has not proved successful, our forays into C–C and C–O activation in molecules with embedded directing groups have provided new chemical knowledge. Our future plans are to return to the 2-amino pyridine system, now armed with the knowledge of what C–C activation can accomplish with 8-acyl quinolines.



