Reports: G3
48059-G3 Aqueous Metal-Ligand Bifunctional Hydrogenation of Carbon Dioxide
Our research group is interested in homogeneous catalysis using transition metal complexes that utilize non-covalent ligand interactions with substrate molecules as their key mechanistic feature. We are inspired by the metal-ligand bifunctional hydrogenation of polar double bonds described by Noyori which is the model for well defined multi-functional organometallic catalysis. Our group has on-going projects involving ligand effects in the metal-ligand hydrogenation reactions of ketones using aqueous and immobilized catalysts, as well as, applications of this reaction to normally non-reactive substrates, such as amides. Under the ACS PRF grant # 48059-G3 we initiated a specific project to explore the catalytic hydrogenation of CO2 to formic acid. We proposed that the hydrogenation of CO2 can occur by a similar concerted transition state to the one described for the metal-ligand hydrogenation of ketones; this hypothesis is supported by theoretical studies on CO2 hydrogenation in a related system.
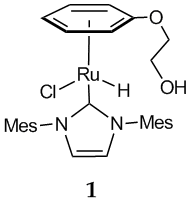
The conventional route to the large class of piano stool (h6-arene)ruthenium(II)L3 complexes, such as 1, involves the intermediacy of the dinuclear [(h6-arene)ruthenium(II)Cl2]2 compounds. The synthesis of these ubiquitous chloro-bridged diruthenium complexes was first described by Winkhaus and Singer in 1967 and has not changed ever since. This involves the reaction of a 1,3 or 1,4 cyclohexadienes with RuCl3.xH2O in ethanolic solution. For our library synthesis the requisite use of ethanol as a solvent in this reaction resulted in mixtures of products that were not resolvable by any means. We began to question the mechanism of the reaction and this prompted us to undertake a detailed examination of this reaction in order to better understand the role of the reagents and solvents. We believe that understanding the mechanism of this reaction can potentially lead to improved synthesis of [(h6-arene)ruthenium(II)Cl2]2 complexes and expand the application of this durable reaction to new generations of piano stool ruthenium compounds. Although this derailed our library synthesis approach, it did lead us to a new avenue of investigation. The work on the synthesis of compound 1 was initiated by Ms. Monique Koppel and was supported by this PRF grant; however, an ARCS Foundation Scholarship is currently supporting Monique in the new area of research involving the nature of the reaction of cyclohexadienes with RuCl3.xH2O.
In a separate study aimed at understanding ligand effects in the hydrogenation of CO2, Mr. Mengping Zhu undertook an examination of substituent effects on the microscopic reverse of the CO2 hydrogenation reaction. We used a substantial part of the year-one PRF funds to purchase a medium pressure Parr reactor to study the CO2 hydrogenation reaction. This reactor includes a custom made catalyst delivery device designed to introduce a small bolus of catalyst solution at the reaction pressure and temperature in order to produce a well-defined zero time point for our kinetic studies and to avoid complications due to ramp-up effects. While we were acquiring, setting up and safety checking our reactor, we decided to go forward with a study of the transfer hydrogenation reaction of ketones using formic acid or the formic acid/triehtylamine azeotrope as the reducing source. The understanding of the reverse of the CO2 hydrogenation reaction has important implications to the forward reaction of interest.
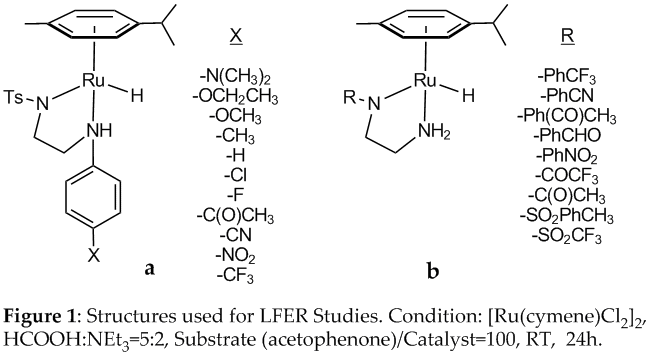 To better understand ligand effects on the
transfer hydrogenation reaction using formic acid we synthesized two ligand
series shown in Figure 1 and
evaluated their relative catalytic activity in the HCO2H/Et3N
azeotrope (reaction conditions shown in the caption). The series in Figure 1a involves systematic variation
of electron releasing and withdrawing groups of the amino (-NH proton donor)
group of the classic Noyori metal-ligand bifunctional catalyst; the series in Figure 1b includes structural
modifications at the amido (non-proton donor). Figure
1 shows the putative hydride catalyst under reaction conditions; we have not
yet isolated any of the hydride catalysts or their chloride pre-catalysts, but
plan to do so in the second year of the grant period.
To better understand ligand effects on the
transfer hydrogenation reaction using formic acid we synthesized two ligand
series shown in Figure 1 and
evaluated their relative catalytic activity in the HCO2H/Et3N
azeotrope (reaction conditions shown in the caption). The series in Figure 1a involves systematic variation
of electron releasing and withdrawing groups of the amino (-NH proton donor)
group of the classic Noyori metal-ligand bifunctional catalyst; the series in Figure 1b includes structural
modifications at the amido (non-proton donor). Figure
1 shows the putative hydride catalyst under reaction conditions; we have not
yet isolated any of the hydride catalysts or their chloride pre-catalysts, but
plan to do so in the second year of the grant period.
When the logarithm of the relative turnover frequencies for the hydrogenation of acetophenone in the HCO2H/Et3N azeotrope using the series in Figure 1a were plotted against the Hammett substituent constants (s) we observed a large and negative reaction constant, r = -4.31. We interpret this as evidence for rate-determining catalyst regeneration involving an ion pair, where the amine ligand is substantially protonated by formic acid (manuscript in preparation). With respect to CO2 hydrogenation, our results are encouraging because they indicate that the reverse reaction (HCO2H dehydrogenation) is slow relative to hydrogenation. This also suggests that CO2 hydrogenation likely occurs by a stepwise ionic mechanism, rather than the concerted one we had originally hypothesized. We are repeating these reactions to extract specific reaction rate constants. In addition, we are currently performing the reactions using the series in Figure 1b.
The research supported by the PRF grant and is currently the focus of a manuscript in preparation. The results of this project have been disseminated at the Inorganic Reaction Mechanisms GRC (March 2009), The Inorganic Chemistry GRS and GRC (June 2009), the 238th ACS National Meeting (August 2009).



