Reports: AC3
47249-AC3 Fundamental Chemistry of New Group 5 Metal Imido and Bis-Imido Complexes
The introduction of the BDI ligand onto niobium was accomplished via salt metathesis with an imidoniobium precursor. We believe that the imido group is particularly useful for forming this product as it both reduces the required coordination number about the metal and supports the d0 oxidation state through its strong donor capability. A number of halide, alkyl, and aryl complexes were characterized, that serve both as starting points for future investigations and as bases of comparison for the electronic properties of the metal center and the nature of BDI-metal bonding interactions.
A second imido group was introduced onto the metal via a salt metathesis reaction. The bis(imido) complex that formed represents a rare example of this ligand set on a Group 5 metal. The complexes were found to lose the coordinated donor ligand, probably through a dissociative mechanism, allowing for pyridyl ligand abstraction by Lewis acids to generate the four coordinate bis(imido) complex (BDI)Nb(NtBu)2 in solution. If left unsaturated, this species underwent a rearrangement to form a cyclo-bis(anilide) niobium complex, in which a hydrogen had been transferred to one of the imido nitrogens. This reactivity supports the conjecture that forming heavily π-loaded systems creates metal centers with more highly polarized M-Lπ bonds. The imido group in this case activates a CH bond, albeit presumably intramolecularly. Cycloaddition of tBuNCO was found to be competitive with the rearrangement process; the metallacycle could be observed spectroscopically and was found to yield tBuNCNtBu on thermal decomposition.
The close energetic proximity of diverse reaction pathways for second-row transition metals can impede progress in research focused on step-wise synthetic chemistry with these metals. However, the results described in this chapter indicate that it is possible, at least in our niobium system, to use the small energy differences associated with the transition states leading to these different products to choose a specific product mixture based on very subtle modifications to the reaction conditions. The specific transformations discussed here have allowed us to access the products of i) metal-based reduction to furnish a stable Nb(III) species, ii) carbon-carbon double bond formation via oxycarbene coupling, and iii) a CH activation product formed by ring-opening of a high-energy metallaoxirane intermediate.
If it proves to be general, this technique could potentially be useful in employing CO to generate different organic species from a single precursor. In addition, the reaction scheme leading to the observed products in our Nb system was mapped by the synthesis of stable analogs to the proposed intermediates, affording us a great deal of detail about the mechanisms leading to this array of products. Additional research should be directed toward uncovering factors that would further improve our ability to control product distributions.
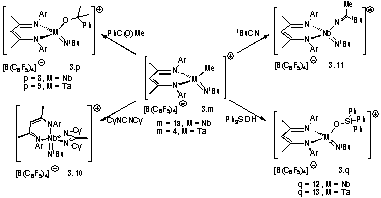
The neutral dimethyl complexes 1.6 (Nb) and 3.3 (Ta) undergo clean methide group protonolysis to yield the stable 12/14 e cations 3.1a and 3.4. These compounds have been thoroughly characterized and, for 3.1a, confirmed structurally. The crystallographic data indicate a shortening of the Nb-Me and Nb=NtBu bonds relative to those observed for 1.6, which is indicative of a complex with both a lower coordination number and higher metal-based cationic charge.
In contrast to the divergent carbonylation chemistry observed with the dimethyl compounds, the methyl cations were not found to irreversibly bind CO. In the presence of phosphines, cationic phosphine-trapped acyl species could be formed cleanly as either a single (PCy3) or a mixture (PEt3) of isomers. When isotopically enriched 13CO was used in the reaction, these complexes were readily identified by diagnostic doublets (1JCP ≈ 50 Hz) in the 13C{1H} and 31P{1H} NMR. The coupling constants and the associated chemical shift ranges of the doublets coincide well with those of analogous complexes.
A related insertion reaction with XylNC was observed to yield a stable cationic iminoacyl species as a mixture of two isomers. This compound was stable in solution up to 398 K and, in contrast to the acyls, did not bind phosphines to yield tetrahedral phosphine-trapped iminoacyls. The relative stability of these compounds, and related iminoacyl complexes as a whole, compared to that of their isoelectronic acyl counterparts has been documented in the literature. The phenomenon is proposed to originate from stronger MN binding compared to the related MO bond of the acyls.
The cationic complexes 3.1a and 3.4 underwent insertion reactions with acetophenone to yield cationic alkoxide complexes, and 3.1a inserted CyN=C=NCy and tBuCN to yield the amidinate and ketimide complexes, respectively. Both the Ta and Nb complexes cleanly extruded methane on reaction with Ph3SiOH to yield the siloxide complexes in high yields.
Finally, while the ethylene polymerization activity of the Ta cation was sluggish, the Nb complex 3.1b afforded high density polyethylene in high yield at room temperature under 1 atm of pressure. The cation failed to yield appreciable quantities of poly(α-olefins) when polymerization reactions were screened under similar conditions with both branched and linear α-olefins, indicating that the steric requirements of the BDI ligand allow only the smallest substrates to enter the coordination sphere of the metal. This is consistent with our original hypothesis regarding the relative inertness of the dimethyl compound 1.6.



