Reports: B3
44534-B3 Sites of Protonation and Ligand Migration in Bimetallic Organometallic Complexes
This report focuses on a computational chemistry study of some bimetallic organometallic complexes. Our computational modeling is closely connected to the experimental work performed in the laboratories of Dr. M. Cowie at the University of Alberta, Edmonton.
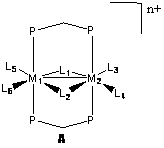
The purpose of this work is to examine fundamental catalytic conversions that occur at the bimetallic centers. The general structure is shown below in A, where the metal centers are from groups 8 and 9 in the periodic table. Though not involved in the chemistry in our study, the diphenyldiphosphinomethane ligand (dppm) provides the structural support necessary to hold the two metals in place. Around the metals are ligands occupying positions L1-L6, involved more directly in this study. Not all of these positions are necessarily occupied at all times, with positions varying based on the system being studied. The ligands involved include acetyl, carbonyl, hydride, methyl, methylene, phosphine, and triflate anion. The overall charge of the complexes includes neutral, cationic, and dicationic, depending on the ligands and metals involved.
Gaussian03 was used for all calculations. The B3LYP theoretical method was employed for all calculations. The cc-pVDZ or cc-pVTZ basis set was utilized for all atoms except the transition metal atoms, for which the LANL2DZ basis set was employed. The dihydrogen diphosphine methane ligand (dhpm) was utilized in place of the dppm ligand for our modeling work.
We report our results on two aspects of this research. The first aspect investigated the migration of a bridging hydride between Rh and Os in the system [RhOs(μ-H)(μ-CH2)(dhpm)2(CO)3(PH3)]2+ (1) which was based on experimental observations by Dr. M. Cowie1.
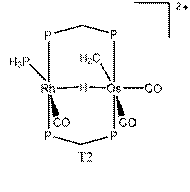
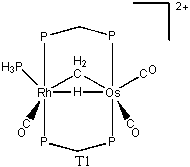
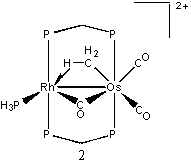
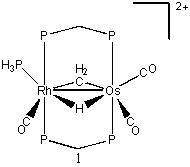
The transition state from 1 to 2 has been difficult to locate. Using QST3, a transition state was thought to be found (T1). This had the hydrogen positioned directly between the metals, with the metals moved apart slightly. The energy of T1 relative to 1 was found to be 40.8 kcal/mol. A frequency was run on this transition state, and three imaginary frequencies were found. Therefore, it is a higher order saddle point and not a true transition state. Further studies were performed at the higher theory level of B3LYP/cc-pVTZ. Upon the completion of a QST3, there was a slightly different transition state (T2). The relative energy of T2 was found to be 36.1 kcal/mol. A frequency run on this new transition revealed that it in fact had only one imaginary frequency. So T2 is a legitimate transition state for this migration. The reaction from 1 → T2 → 2 has relative energies of 6.7, 36.1, and 0.0 kcal/mol, respectively. With zero-point energy corrections at the B3LYP/cc-pVTZ level, the reaction has relative energies of 5.2, 31.6, and 0.0 kcal/mol, respectively.


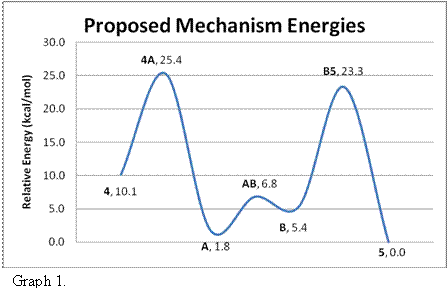
In the second aspect, we studied the reaction of butadiene with (3) the Ir-Ir system [Ir2(CO)2(CH3)(dhpm)2]+. Our focus was on the validity of a proposed reaction mechanism of the reaction of 4 to 5. The mechanism was first proposed by Dr. M. Cowie in one of his articles2 and our investigation considered the relative energy barriers between each proposed step. Structures 3, 4 and 5 were experimentally observed by Cowie, and structures A and B were proposed to be intermediate steps in the reaction of 4 to 5. The relative energies of the various states are displayed in Graph 1.
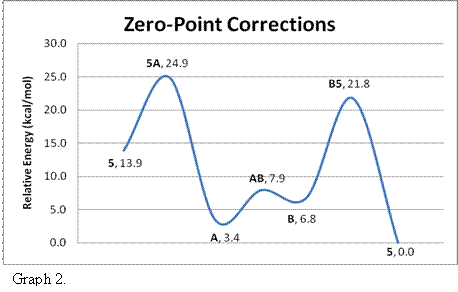
The results with zero point energy corrections are displayed in Graph 2. Given that the energy barriers are all <20 kcal/mol, the proposed mechanism is acceptable. Even the placement of equilibrium between A and B is suitable. Our results support Cowie's mechanism.
References
[1] Bridging Methylene and Methyl Complexes of Rh/Os: Influence of the Ancillary Ligands on the Methyl Binding Mode, Wigginton, J. R.; Trepanier, S. J.; McDonald, R.; Ferguson, M. J.; Cowie, M., Organometallics, 2005, 24 , 61946211.
2 Double Activation of the Geminal Carbon-Hydrogen Bonds in 1,3-Butadiene by a Diiridium Complex, Ristic-Petrovic, D.; Torkelson, J. R.; Hilts, R. W.; McDonald, R.; Cowie, M. Organometallics 2000, 19, 4432-4434.



