ACS PRF | ACS
All e-Annual Reports

41883-B1
Strategy for the Enantioselective Preparation of Polyhydroxy Pyrrolidines: Synthesis of DAB-1 and (-)-Anisomycin
Our research reports to date have focused on a novel synthetic strategy that provides stereo- and enantiocontrolled access to significant polyhydroxylated pyrrolidines such as anisomysin and DAB-1. In this edition, however, we disclose our recent efforts to extend this methodology from the five-membered pyrrolidine ring system to the six-membered piperidine ring system. This family of compounds exhibits a wide array of interesting biological activities, which has placed piperidines under the close scrutiny of the pharmaceutical industry. We showcase our new strategy for the preparation of these six-membered heterocycles with the synthesis of coniine, a poisonous alkaloid that is notorious for being the causative agent in spotted hemlock that took the life of Socrates.
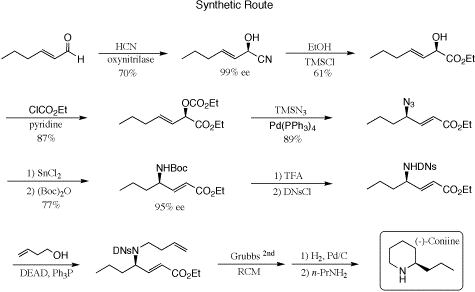
Our enantiospecific route is outlined in the reaction scheme below. Induction of asymmetry (>99% ee) in our achiral starting material, 2-hexenal, was achieved using the almond enzyme oxynitrilase, and denotes a key step in this synthesis. Hydrolysis of the product (R)-cyanohydrin with catalytic TMSCl in dry ethanol followed by a cold quench with an equivalent of water produced the corresponding ethyl ester without observable racemization. The alpha-hydroxy ester was then converted into the allylic ethyl carbonate in preparation for the crucial 1,3-substitutive chiral transfer step. To our delight, exposure of the allylic carbonate and trimethylsilylazide to tetrakis(triphenylphosphine)palladium(0) catalyst provided the rearranged azido product with complete stereo- and regiochemical control, another highlight of this work. A sequential one-pot reduction of the (R)-azido group with stannous chloride followed by protection with di-tert-butyldicarbonate produced the desired Boc-protected gamma-amino ester in 77% yield. Acid-catalyzed removal of the Boc group and subsequent reaction with Moshers acid chloride afforded a diastereomeric mixture that established the enantiomeric excess at 95%, as measured by fluorine NMR. Reaction of this same amino function with 2,4-dinitrobenzenesulfonyl chloride (DNsCl) produced the analogous sulfonamide. This activating group was necessary to effect the clean N-alkylation of 3-buten-1-ol under Mitsunobu conditions in the ensuing step. Finally, ring-closing metathesis (RCM) on the diene using the Grubbs second-generation catalyst led to DNs-protected coniine. The DNs group is easily removed under mild conditions with excess n-propyl amine. We are currently optimizing the yields on the latter synthetic steps in preparation for publication.