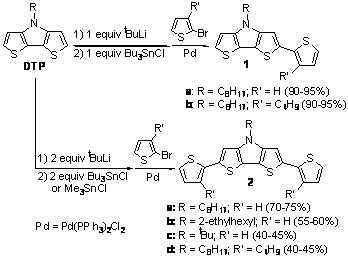ACS PRF | ACS
All e-Annual Reports

41750-AC7
Thiophene Amination Routes to Nitrogen Functionalized Conjugated Polymers
ACS-PRF Narrative Report - September 3, 2007
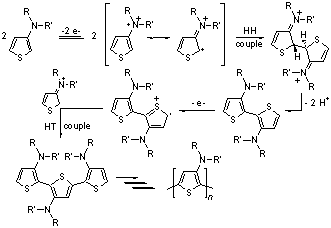
Since our last report, we have continued to make advances on various aspects of the proposed project. We have continued to study the polymerization mechanism of N-alkyl-3-aminothiophenes and their derivatives. Such alkylaminothiophenes exhibit two oxidations, theorized to be first oxidation of the nitrogen at ~0.5 V, followed by oxidation of the thiophene ring at ~1.8 V. Investigation of quatenary ammonium analogues have shown that be removing the oxidizable nitrogen lone pair, these monomers exhibit only a single oxidation peak at +1800 mV, verifying the nitrogen as the initial oxidation. Investigation of the first oxidation in more detail, it has been shown to be electrochemically irreversible, but chemically reversible and fast scanning can allow oxidation and re-reduction to regenerate the initial alkylaminothiophene without the detectable formation of dimer or oligomer formation. However, holding the potential for prolonged periods between the two observed oxidations does result in normal alpha-alpha coupled oligomer and polymer formation, thus suggesting that the initial nitrogen-based oxidation can result in thiophene-based radicals via resonance. Calculations (HF 6-31G*) show HOMO contributions on both the amine nitrogen and thiophene ring, and spin density calculations of the corresponding radical cation show equal spin contributions on both the nitrogen and the 2-position of the thiophene ring. In contrast, little to no spin density is observed at the 5-position, supporting the high regioregularity observed in the resulting polymers (85-90%). Our combined electrochemical and computational studies have resulted in the proposed electropolymerization mechanism illustrated in Scheme 1.
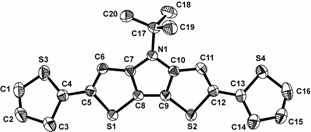
Scheme 1. Proposed electropolymerization mechanism for aminothiophenes As previously reported in Organic Letters, we had successful prepared dithieno[3,2-b:2',3'-d]pyrroles (DTP)-based oligothiophenes that exhibit enhanced fluorescence efficiencies in solution. Last year we had begun investi-gation of side chain effects with the preparation a new N-t-butyl analogue (2c) and have since continued this study. While the fluorescence intensities of the DTP-based quaterthiophenes 2a-d are significantly enhanced in comparison to the parent quaterthiophene (quantum efficiencies of 36-66 % in cyclohexane), it was found that the alkyl groups add high frequency modes that increase with the number of side chain methylene units. This increase in high frequency modes results in a greater number of deactivation pathways via internal conversion and an overall decrease in fluorescence efficiency. This effect can be seen in 8a-c, where N-functionality is varied from the simple octyl side chain to the branched 2-ethylhexyl, and finally to fully branched tert-butyl group. As the number of methylene units decreases in this series (7-5-0), a significant increase can be observed in the fluorescence efficiency (30-47-65% in CH3CN and 49-60-66% in cyclohexane). Scheme 2. Synthesis of Dithieno[3,2-b:2',3'-d]pyrrole Oligomers Of greater interest, however, was whether the crystal packing and solid-state photophysics could be tuned via side chain induced sterics. Shortly before last year's report, we had successfully crystallized 2c and begun initial structural studies. Since then we have optimized the refinement to successfully give the first such structure of a DTP-based oligothiophene (Figure 1) beyond that of the core DTP unit itself. Figure 1. Ellipsoid plot of oligomer 2c at the 40% probability level. The diffraction patterns of thin films of 2c were also obtained in order to compare the packing of the processed thin films to the single crystal data. The primary diffraction spacing of the films was found to be in relatively good agreement with the single crystal data. For a more absolute comparison, the single crystal data was then used to calculate its corresponding theoretical powder diffraction pattern. Comparison of the experimental thin film and calculated powder patterns resulted in nearly perfect agreement, strongly supporting that the solution-processed thin film retains the packing exhibited by the single crystal. For comparison, the thin films of 2a and 2b were also investigated to reveal smaller d-spacings consistent with the tighter packing of the oligomers, as might expected as a result of the reduced sterics of the N-functionalized side chains. The solid-state emission spectra for the DTP-based oligothiophenes 2a-c are shown in Figure 2. While the measurements of the solid-state emissions were not optimized to acquire the maximum emission intensities, measurements performed under identical conditions showed a significant difference in emission intensity with the varied N-functionalization of the DTP unit. The octyl side chain of 2a resulted in films with very weak, but measurable emission. The use of branched side chains, however, resulted in an increase in emission intensity, with the greatest intensity produced from the use of the tert-butyl side chain in 2c. This trend agrees with previously discussed thin film diffraction data and strongly supports the idea of tuning of the solid-state emission via modification of the DTP side chain sterics. A full paper describing the solution and solid-state properties of these DTP-based oligomers is currently under review at Chemistry of Materials. Figure 2. Solid-state absorption and emission spectra of 2a-c