Reports: B1
48122-B1 Cycloaddition Reactions for the Formation of Indolines, Indoles and Benzofurans, and Subsequent Functionalization Reactions
Our Research report focuses on the accomplishments achieved during this first year; this work has been divided into two different parts:
-The first one presents a novel oxidative [2+3] cycloaddition process, between a substituted phenol and an alkene, allowing to generate, in only one step, different heterocyclic rings such as dihydrofuranobenzofurans, tetrahydrofuranobenzofurans, tetrahydropyranofurans and dihydrobenzofurans. Formally, this transformation may be termed as an "aromatic ring umpolung". Whereas an electron-rich aromatic nucleus, such as a phenol, normally reacts as a nucleophile, suitable oxidative activation can convert it to a reactive electrophilic intermediate, such as a phenoxonium ion. The high electronegativity of the oxygen atom makes this transient intermediate short-lived, excessively electrophilic and consequently very reactive; it may then be intercepted by external mild nucleophiles such as an electron-rich double bond, Scheme 1.
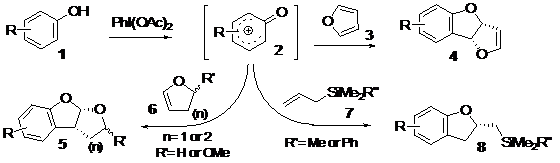
The formation of the heterocylclic core would be produced in two steps; first, the electrophilic species 2 would be trapped by the electron-rich double bond in a manner consistent with electrophilic substitution/addition rules to generate the species 10, which would be secondly trapped by a nucleophilic attack of the hydroxyl group present on 10 to generate the ring 11. That is the reason why we assimilate this process to a formal [2+3] cycloadition, Scheme 2.
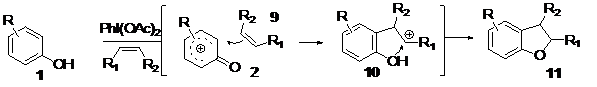
Using this method, numerous and different heterocyclic cores have been generated. In addition, this transformation is applicable to a large range of phenols, and can tolerate a hindered group in ortho-position to the hydroxyl. However, the reaction with furan is sensitive to the stereoelectronic effect that could be generated by a methoxy group in meta-position. These reactions, leading to different heterocyclic compounds, could be the key to the concise syntheses of numerous natural compounds containing such heterocyclic rings, such as Panacene, Psorofebrin, Tenuisine C, Aflatoxin B, etc. As an illustration, we have performed a concise diastereoselective synthesis of Panacene, a shark antifeedant compound, in only five steps. In addition, this synthesis has allowed us to develop an unknown and diastereoselective method to produce the bromo-allene moiety efficiently. We envisage developing novel applications in total synthesis of this transformation in the future, Scheme 3.
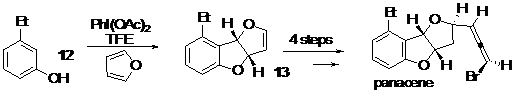
-The second part describes a novel metal-free and environmentally benign method for the formation of C-C bonds between dienimides and furan derivatives to quickly produce aryl-aryl or aryl-alkyl coupling, Scheme 4.
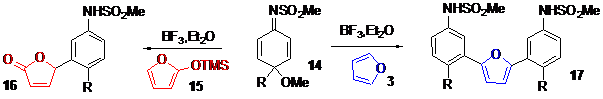
This process occurs with unactivated compounds via a Michael-rearomatization tandem process in good yields, and only requires the elimination of methanol as a by-product. This reaction carried out on different dienimides, tolerates different functionalities, such as a protected alcohol, a mesylate, a free alcohol or an ester on the aliphatic side chain, as a spectator functional groups. A description of a study, demonstrating that dienimide is an efficient Michael acceptor to accomplish such transformation is described, and explains why this method is not efficient with similar dienone compounds, Scheme 5.
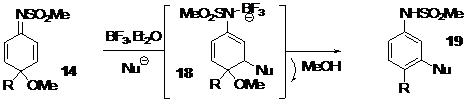
In
addition, this reaction in presence of furan has yielded an intriguing side
product, in a 13% ratio, containing an exotic pentacyclo[5.4.0.0.0.0]undecane core, 23.
The formation of this birdcage structure can be explained if we consider that a double formal
cyloaddition tandem process has occurred. Indeed, we
assume that intermediate 20 is trapped by the mesyl-enamide
group to generate 21 via a first formal [4+2] cyloaddition.
The Lewis acid should activate the remaining enimide
functionality that would be trapped by the double bond of the dihydrofuran moiety to generate species




