Reports: AC1
47272-AC1 Short, Tunable Chiral Peptidic Ligands for Osmium Tetroxide-Mediated Chemistry
We now report the discovery of an organocatalytic enantioselective protocol for the chlorolactonization of 4-substituted 4-pentenoic acids
Table 1. The (DHQD)2PHAL mediated halolactonization.
|
Entry
|
X+ Sourcea
|
Solvent
|
Additive
|
ûC
|
% eeb
|
|
1
|
NBS
|
CH2Cl2
|
-
|
RT
|
22
|
|
2
|
NBS
|
CH2Cl2
|
-
|
-20
|
14
|
|
3
|
NBS
|
CHCl3
|
-
|
RT
|
35
|
|
4
|
NCS
|
CH2Cl2
|
-
|
RT
|
39
|
|
5
|
NCS
|
CHCl3
|
-
|
RT
|
65
|
|
6
|
NCS
|
CHCl3
|
-
|
-40
|
NRc
|
|
7
|
DCDMH
|
CHCl3
|
-
|
RT
|
71
|
|
8
|
DCDMH
|
CHCl3
|
-
|
-40
|
83
|
|
9
|
DCDMH
|
CHCl3
|
PhCO2H
|
-40
|
86
|
|
10
|
DCDMH
|
CHCl3/Hex (1:1)
|
-
|
-40
|
86
|
|
11
|
DCDMH
|
CHCl3/Hex (1:1)
|
PhCO2H
|
-40
|
89
|
|
12
|
DCDPH
|
CHCl3/Hex (1:1)
|
PhCO2H
|
-40
|
89
|
aX = Br for entries 1-3, and Cl for entries 4-12. bas judged by chiral HPLC or GC analysis. cNR = no reaction after 3 hours.
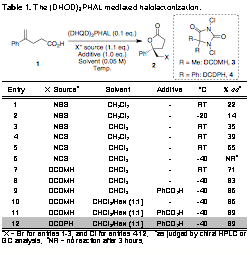
We were encouraged by the discovery that (DHQD)2PHAL provided good conversions and modest enantioselectivity in the bromolactonization of alkenoic acid 1 (Table 1, entry 1). When 1 was treated with 10 mol % of (DHQD)2PHAL in the presence of NBS in CH2Cl2, the desired bromolactone was isolated in quantitative yield with a modest 22% ee. Although lowering the reaction temperature to -20 ûC produced an inferior result (entry 2), an exhaustive solvent screen subsequently revealed that CHCl3 was optimal, returning the bromolactone with an improved selectivity of 35% ee (entry 3).
A substantial improvement in selectivity was realized when the analogous chlorolactonization was investigated, employing NCS in lieu of NBS. In this case, reaction with 10 mol % of catalyst in CHCl3 returned the desired chlorolactone with a dramatically enhanced selectivity of 65% ee (entry 5). Analogous to the bromolactonization, chlorolactonization in DCM returned the chlorolactone with an eroded 39% ee (entry 4).
| Scheme 1. Cyclization of 5f with various N-chlorohydantoins.
|
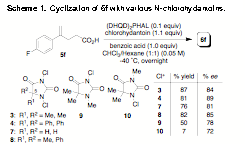
Although we were initially discouraged by the sluggish chlorolactonization when the temperature of the reaction was lowered to -40 ûC (no lactone was detected by TLC, entry 6), we were emboldened by the discovery that the more reactive 1,3-dichloro-5,5-dimethylhydantoin (3, DCDMH) afforded a faster and more stereoselective reaction at -40 ûC (entry 8, quant. conversion, 83% ee; for absolute stereochemical assignment of product see Supporting Information). The reaction at room temperature was less stereoselective (entry 7). Extensive parallel additive and co-solvent screens revealed that both 1.0 equiv of benzoic acid additive (entry 9) and a CHCl3/hexane (1:1) solvent system (entry 10) each independently increased the selectivity of the chlorolactonization to 86% ee. When the co-solvent and additive effects were combined, the desired lactone was produced in 89% ee (entry 11). This selectivity was maintained when the analogous DCDPH (4) was applied as the terminal chlorine source (entry 12). We deferred to DCDPH, since it returned higher isolated yields during the course of scale-up experiments. Importantly, it appears that the N,N-dichlorohydantoins are uniquely situated between NBS and NCS in reactivity. They are reactive enough relative to NCS to allow for reaction at low temperature yet are attenuated relative to NBS, thus quelling a prevailing non-selective background reaction.
Next, a panel of p-substituted pentenoic acids was subjected to the optimal reaction conditions (Table 2). Phenyl substituted lactone 6a was generated in 86% yield and 89% ee. The quasienantiomeric (DHQ)2PHAL returned the enantiomeric lactone 6b in a reduced 75% yield and 77% ee. The reduced selectivity in the latter case is likely due to the diastereomeric relationship between the DHQD and DHQ catalysts. Lactone 6c was returned in nearly quantitative yield, but with essentially no selectivity. Evidently, the p-methoxy substituent promotes the facile ring opening of the putative chloronium intermediate, precluding a high degree of selectivity. Lactone 6d, harboring a less donating p-methyl group, was returned with a much improved 80% ee. Chloro and fluoro substituted lactones 6e and 6f were generated in good yield with 88% and 89% ee, respectively. Para-trifluoromethyl substituted 6g was isolated with 90% ee. Biphenyl substituted substrate 5h returned chlorolactone 6h in 83% ee. Larger aryl substituents resulted in reduced selectivities. Namely, 2-naphthyl lactone 6i was returned in 72% ee. Finally, replacing the 4-aryl substituent in 5 with a cyclohexyl group returned lactone 6j in 55% yield with a substantially lower 43% ee.
Table 2. (DHQD)2PHAL mediated asymmetric chlorolactonization.
|
Rb
|
%yieldc
|
%eed
|
Rb |
%yieldc
|
%eed
|
|
Ph; 6a
|
86
|
89
|
p-F-C6H4; 6f
|
81 (78)
|
89 (86)
|
|
Ph; 6b (ent-6a)
|
75
|
77e
|
p-CF3-C6H4; 6g
|
61
|
90
|
|
p-OMe-C6H4; 6c
|
99
|
<5
|
p-Ph-C6H4; 6h
|
59
|
83 (80)
|
|
p-Me-C6H4; 6d
|
86 (82)f
|
80 (82)f
|
2-Napth; 6i
|
92 (83)
|
72 (72)
|
|
p-Cl-C6H4; 6e
|
80
|
88
|
Cy; 6j
|
55
|
43
|
aReaction times; 30 min for products 6a-6f and 6j, 90 min for products 6g-6i (as judged by TLC). bStereochemistry was determined by chemical correlation (see Supplementary Information). cIsolated yield after column chromatography. dAs judged by chiral GC or HPLC analysis. eReaction was performed with 0.1 equiv of (DHQ)2PHAL. fValues in parentheses are yields and % ee when 0.01 eq. (DHQD)2PHAL was employed.
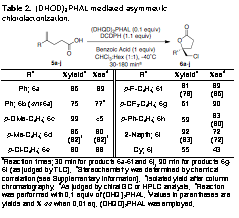
We have also evaluated the transformation when 0.01 equiv of (DHQD)2PHAL was employed for a selection of the substrates in Table 2. Even with a low catalyst loading of just 1 mol %, lactones 6d, 6f, 6h, and 6i were returned in 82%, 86%, 80%, and 72% ee, respectively (Table 2, values in parentheses). In each case the enantioselectivities approximately match those realized with 10 mol % catalyst.
| Scheme 3. One-pot conversion of 6f to chiral epoxyalcohol 12.
|
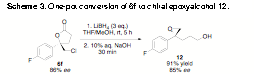 We have developed a convenient one-pot protocol for
the transformation of these chiral chlorolactones into enantioenriched
1,1-disubstituted epoxy alcohols (Scheme 3). Lithium borohydride reduction of lactone 6f (86% ee)
followed by sodium hydroxide mediated cyclization of the resulting chlorohydrin
intermediate returned 1,1-disubstituted epoxy alcohol 12 in good yield and importantly without any appreciable
loss of enantiopurity.
Significantly, 4-arylpenten-1-ols have proven to be difficult substrates
for conventional asymmetric epoxidation protocols.
We have developed a convenient one-pot protocol for
the transformation of these chiral chlorolactones into enantioenriched
1,1-disubstituted epoxy alcohols (Scheme 3). Lithium borohydride reduction of lactone 6f (86% ee)
followed by sodium hydroxide mediated cyclization of the resulting chlorohydrin
intermediate returned 1,1-disubstituted epoxy alcohol 12 in good yield and importantly without any appreciable
loss of enantiopurity.
Significantly, 4-arylpenten-1-ols have proven to be difficult substrates
for conventional asymmetric epoxidation protocols.
In summary, we have discovered a novel organocatalytic asymmetric chlorolactonization that returns chiral chlorolactones by action of (DHQD)2PHAL and DCDPH. This methodology represents the first example of a catalytic, enantioselective halolactonization that proceeds with synthetically useful enantioselectivities.



