Reports: G1
46550-G1 The Development of Mild and Operationally Simple Direct Methods for the Synthesis of beta-Dicarbonyl Compounds
Soft enolization has been shown to provide an exceptionally mild and operationally simple approach to direct carbon-carbon bond formation applicable to 1,3-diketone and b-keto thioester synthesis.
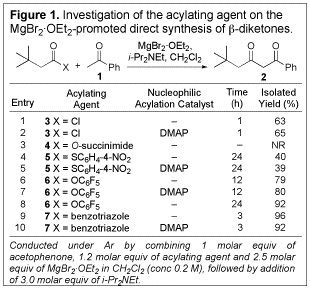
1,3-Diketone synthesis. To explore the use of soft enolization in 1,3-diketone synthesis, acetophenone was combined with a variety of known acylating agents both with and without added DMAP as a nucleophilic acylation catalyst. The results are summarized in Figure 1. Addition of DMAP was uniformly of no benefit with regard to either the time required for the reaction or the yield produced (entries 2, 5, 7, and 10). O-Succinimide ester 4 failed to react altogether, and while thioester 5 did produce the desired product, yields were lower than for the corresponding acid chloride (3). O-Pfp ester 6 proved to be a suitable acylating agent, giving 79% yield of the b-diketone within 12 h and 92% within 24 h. Even better yields and shorter reaction times resulted from the use of N-acylbenzotriazole 7. Thus, both O-Pfp esters and N-acylbenzotriazoles were investigated in our subsequent studies.
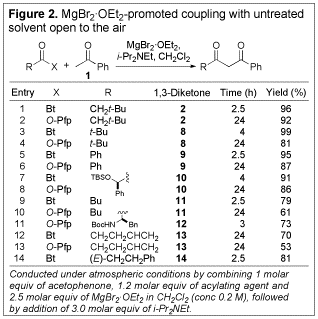
Having secured a mild and straightforward method for the synthesis of b-diketone 2, we determined the scope of the method with respect to other N-acylbenzotriazoles and O-Pfp esters (see Figure 2). In general, the N-acylbenzotriazoles outperformed the O-Pfp esters in terms of both reaction time and yield. The isolated yields were typically excellent when N-acylbenzotriazoles were used. Significantly, the coupling reaction could be carried out in the presence of an acidic urethane nitrogen protecting group (entry 11), and also in the presence of an enone (entry 14), without detrimental results, as would be expected in the corresponding hard enolization-based processes.
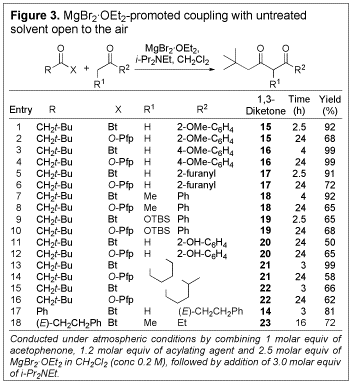
We explored the scope of the coupling reaction using a variety of ketones with various N-acylbenzo- triazoles and O-Pfp esters (see Figure 3). Once again, in all cases the desired 1,3-diketone was obtained in good to excellent yield. Notably, the coupling could be conducted with cyclohexanone as the nucleophile to give the corresponding mono-substituted 1,3- diketone (21) in excellent yield (entry 13). Entries 11 and 12 reveal that the process is even compatible with the presence of phenolic functionality. Such a substrate would not be amenable to traditional coupling methods without prior incorporation of a phenol protecting group. A significant result is shown in entry 18 where 1-[(E)-cinnamoyl]-1H-benzotriazole and 3-pentanone could be coupled to give the desired 1,3-dicarbonyl (23) without subsequent cyclization to the corresponding 1,3-cyclohexanedione, as is typical of such systems.
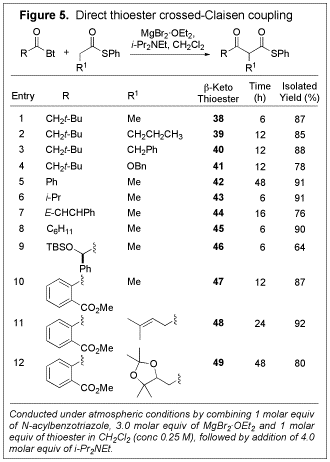
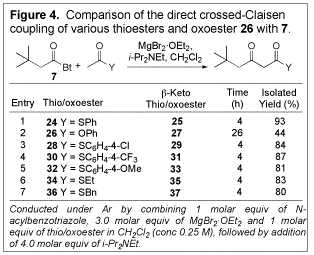
Crossed-Claisen condensation. We anticipated that the use of a thioester as the enolate precursor, in combination with an acid chloride or N-acylbenzotriazole as an acyl donor would enable chemoselective enolization leading to a controlled direct crossed-Claisen coupling. To test this idea, N-acylbenzotriazole 7 and thioester 24 were combined in CH2Cl2 in the presence of MgBr2áOEt2 and Hunig's base (Figure 4), and the desired product was obtained in excellent yield (93%). Neither the self-addition products nor the other crossed-Claisen products were detected. To confirm the importance of the thioesters in this transformation, oxoester 26 was treated under analogous conditions. In this case, only a relatively low yield (44%) of b-keto oxoester (27) formed after an extended period of time, thus confirming the superiority of thioesters in the transformation. Different thioesters were evaluated for their effect on this coupling reaction; S-phenyl thioester (24) was found to be the best. The transformation could be conducted using untreated, reagent grade open to the air.
Having
established proof of concept in the direct thioester crossed-Claisen coupling
and confirming that it could be conducted without the need for highly controlled
conditions, we investigated its scope (Figure 5). In general, the
transformation proceeded very well with a range of thioesters and N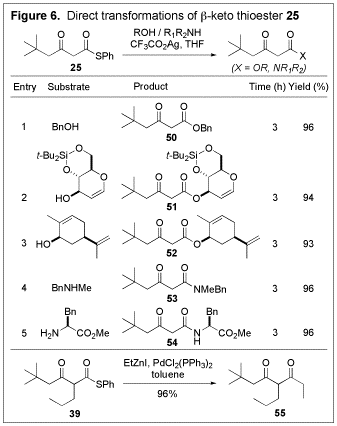
Due to the
unusual reactivity of thioesters, the 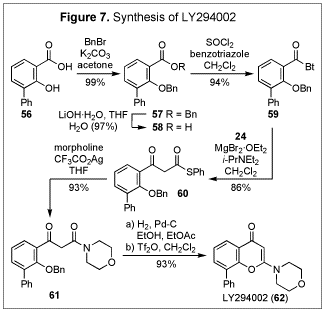
The usefulness of the present method in preparing b-keto thioesters, along with the strategic advantage presented in their synthetic equivalence to b-keto acids, was demonstrated by a concise total synthesis of LY294002 (62) (Figure 7), a potent and specific inhibitor of PI3-K.



