Reports: G3
45685-G3 An Electrochemically Controlled Separation of Nitrogen and Sulfur Heterocycles from Crude Petroleum by Reversible Binding to Iron Bis(dithiolene) Complexes
The research supported by this PRF grant broadened from its initial focus of reversible binding of ligands to iron and cobalt dithiolene complexes to include a systematic exploratory synthesis of multimetallic dithiolene complexes and their properties. This synthetic work focused on the group 10 metals nickel, palladium and platinum because of the nearly ideal square planar corrdination geometries these metals adopt with dithiolene ligands. The fundamental motivation for this work was an interest in further developing access to and understanding of systems capable of supporting multiple electron transfer processes.
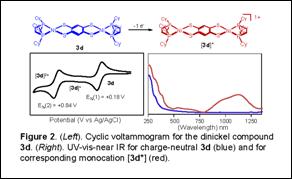
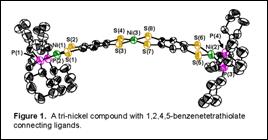
Dimetallic compounds [(P-P)M(S2C6H2S2)M(P-P)] (M = Ni, Pd; P-P = chelating bis(phosphine), 3a-3f) were prepared from O=CS2C6H2S2C=O or nBu2SnS2C6H2S2SnnBu2, which are protected forms of 1,2,4,5-benzenetetrathiolate, a type of bis(dithiolene) ligand. Selective monodeprotections of O=CS2C6H2S2C=O or nBu2SnS2C6H2S2SnnBu2 led to [(P-P)Ni(S2C6H2S2C=O)] or [(P-P)Ni(S2C6H2S2SnnBu2)]; the former was used to prepare trimetallic compounds [(dcpe)Ni(S2C6H2S2)M(S2C6H2S2)Ni(dcpe)] (M = Ni (6a) or Pt (6b) (Figure 1); dcpe = 1,2-bis(dicyclohexylphosphino)ethane). Compounds 3a-3f are redox active and display two oxidation processes, of which the first is generally reversible (Figure 2). Dinickel compound [(dcpe)Ni(S2C6H2S2)Ni(dcpe)] (3d) reveals two reversible oxidation waves with ΔE½ = 0.66 V, corresponding to Kc of 1.6 x 1011 for the mixed valence species. Electrochemical behavior is unstable to repeated scanning in the presence of [Bu4N][PF6] electrolyte but indefinitely stable with Na[BArF24] (BArF24 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate), suggesting that the radical cation generated by oxidation is vulnerable to reaction with PF61-. Chemical oxidation of 3d with [Cp2Fe][BArF24] led to formation of [3d][BArF24]. Structural identification of [3d][BArF24] revealed appreciable shortening and lengthening of CS and CC bond distances, respectively, within the tetrathioarene fragment compared to charge-neutral 3d, indicating this to be the redox active moiety. Attempted oxidation of [(dppb)Ni(S2C6H2S2)Ni(dppb)] (3c) (dppb = 1,2-bis(diphenylphosphino)benzene) with AgBArF24 produced [[(dppb)Ni(S2C6H2S2)Ni(dppb)]2(µ-Ag)2][BArF24]2, [4c][BArF24]2, in which no redox chemistry occurred. Near IR spectroscopy upon cationic [3d]+ (Figure 2) and neutral 6a revealed multiple intense absorptions in the 950-1400 nm region. Time-dependent DFT calculations on a 6a model compound indicated that these absorptions are transitions between ligand-based π-type orbitals that have significant contributions from the sulfur p orbitals.
Tetraphosphinobenzene-linked dimetallic compounds with dithiolene capping ligands were prepared in yields of 60-70% from the corresponding dimetal tetrachloro compounds by transmetalation reactions with dialkytin protected forms of the dithiolene ligands (Method 1, Scheme 1). We observed this approach to be generally cleaner and higher yielding than an alternative route proceeding by halide displacement from X2M(tpbz)MX2 (X = halide; tpbz = 1,2,4,5-tetrakis(diphenylphos-phino)benzene) with fully reduced ene-1,2-dithiolate salts (Method 2, Scheme 1). The resulting dimetal compounds are air-stable and soluble enough to be readily crystallized and interrogated by a variety of physical methods. Related monometallic dithiolene bis(phosphine) compounds with the 1,2-bis(diphenylphosphino)benzene ligand (dppb) were similarly synthesized.

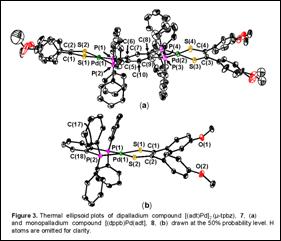
Diffraction quality crystals of the dipalladium
compound bearing the p-anisyl substituted dithiolene ligand (adt) (7),
were grown by vapor diffusion techniques. Compound 7 crystallized with
an unusual asymmetric unit in ![]() consisting of three
independent molecules on general positions and two half-molecules resting on
inversion centers such that Z = 8. The differences between these several
molecules, which presumably obviate a smaller asymmetric unit and higher
symmetry crystal system, are variations in the degree of tetrahedralizing
distortions at the palladium centers and differences in the magnitude of the
folding angle between the mean plane of the central tetraphosphinobenzene
ligand and the pseudo square planar enviroment of each metal atom. One of these
molecules is illustrated in Figure 3 (a). The tetrahedralization at each
metal atom may be quantified by the angle between the S2Pd and P2Pd
planes in the immediate S2PdP2 coordination environment
of Pd. These values range from 3.2°-15.7°, the average being 9.0°. Angles
between the S2PdP2 and P2C6P2
mean planes, which express the magnitude of the chair-type conformation visible
to the molecule in Figure 3, range from 13.4°-31.0°. These deviations from
idealized D2h symmetry contrast with the structure
observed for [(dcpe)Pd(S2C6H2S2)Pd(dcpe)]
(dcpe = 1,2-bis(dicyclohexylphosphino)ethane), which crystallized with near
perfectly planar P2Pd(S2C6S2)PdP2
core. The monopalladium compound [(adt)Pd(dppb)] (8) was also characterized
structurally (Figure 3 (b)) but was observed to have the square planar
coordination geometry that is more typical for Pd(II) in this strong ligand
field environment. Average CS and CCchelate bond lengths for 7
are 1.771[1] and 1.348[3] Å, respectively, values consistent with fully reduced
ligand and a Pd(II) oxidation state.
consisting of three
independent molecules on general positions and two half-molecules resting on
inversion centers such that Z = 8. The differences between these several
molecules, which presumably obviate a smaller asymmetric unit and higher
symmetry crystal system, are variations in the degree of tetrahedralizing
distortions at the palladium centers and differences in the magnitude of the
folding angle between the mean plane of the central tetraphosphinobenzene
ligand and the pseudo square planar enviroment of each metal atom. One of these
molecules is illustrated in Figure 3 (a). The tetrahedralization at each
metal atom may be quantified by the angle between the S2Pd and P2Pd
planes in the immediate S2PdP2 coordination environment
of Pd. These values range from 3.2°-15.7°, the average being 9.0°. Angles
between the S2PdP2 and P2C6P2
mean planes, which express the magnitude of the chair-type conformation visible
to the molecule in Figure 3, range from 13.4°-31.0°. These deviations from
idealized D2h symmetry contrast with the structure
observed for [(dcpe)Pd(S2C6H2S2)Pd(dcpe)]
(dcpe = 1,2-bis(dicyclohexylphosphino)ethane), which crystallized with near
perfectly planar P2Pd(S2C6S2)PdP2
core. The monopalladium compound [(adt)Pd(dppb)] (8) was also characterized
structurally (Figure 3 (b)) but was observed to have the square planar
coordination geometry that is more typical for Pd(II) in this strong ligand
field environment. Average CS and CCchelate bond lengths for 7
are 1.771[1] and 1.348[3] Å, respectively, values consistent with fully reduced
ligand and a Pd(II) oxidation state.
The tpbz ligand has been observed to be an effective insulator between redox active metal centers. Thus, the cyclic voltammetry of 7 was anticipated to reveal a single reversible oxidation wave corresponding to the simultaneous one-electron oxidation of both metallodithiolene end groups. By analogy to the related compound [(Ph2C2S2)Ni(dppe)] (dppe = 1,2-bis(diphenylphosphino)ethane), this oxidation was expected to be an ene-1,2-dithiolate to thienyl radical monoanion oxidation (Scheme 2, (a) → (b)). Surprisingly, the electrochemistry for 7 revealed two well-separated and reversible oxidation waves at 0.00 V and 0.50 V vs Fc+/Fc (Figure 4 (a)). This observation implied either a non-insulating quality to the tpbz ligand or a capacity of the Pd(adt) fragment to sustain a second oxidation such that each end of 7 is doubly oxidized and produces overall a tetracation.
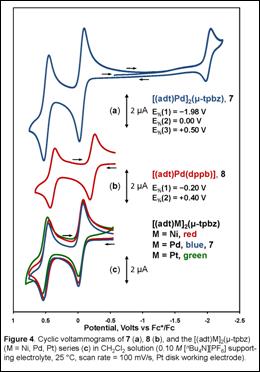 Recourse to
compound 8 was indispensible in interpreting the electrochemical data
for 7. Cyclic voltammetry upon an equimolar solution of 8 under
the same conditions as employed for 7 revealed two reversible oxidations
that were nearly superimposable upon those of 7 but at half the
current amplitude (Figure 4, (b)). This observation proved that 7
is essentially a mere sum of parts (twice 8) While the first of these
oxidations may be plausibly assumed to be dithiolene ligand oxidation ((a)
→ (b) in Scheme 2), the nature of the second oxidation was less immediately
clear. Both Pd(II) to Pd(III) oxidation and thienyl radical monoanion to
dithioketone oxidation ((b) → (c), Scheme 2), which would
mean that the three-member, ligand-based electron transfer series is fully
traversible in this compound, are possibilities. The latter possibility of
completely ligand-based redox chemistry was strongly indicated by a complete
invariance of the voltammetry when the Ni and Pt analogues of 7 were
measured (Figure 4 (c)).
Recourse to
compound 8 was indispensible in interpreting the electrochemical data
for 7. Cyclic voltammetry upon an equimolar solution of 8 under
the same conditions as employed for 7 revealed two reversible oxidations
that were nearly superimposable upon those of 7 but at half the
current amplitude (Figure 4, (b)). This observation proved that 7
is essentially a mere sum of parts (twice 8) While the first of these
oxidations may be plausibly assumed to be dithiolene ligand oxidation ((a)
→ (b) in Scheme 2), the nature of the second oxidation was less immediately
clear. Both Pd(II) to Pd(III) oxidation and thienyl radical monoanion to
dithioketone oxidation ((b) → (c), Scheme 2), which would
mean that the three-member, ligand-based electron transfer series is fully
traversible in this compound, are possibilities. The latter possibility of
completely ligand-based redox chemistry was strongly indicated by a complete
invariance of the voltammetry when the Ni and Pt analogues of 7 were
measured (Figure 4 (c)).



