Reports: AC1
47591-AC1 Asymmetric Hydroamination Catalyzed by Non-Metallocene Complexes of the Group III and Group IV Metals
The intramolecular
hydroamination of carbon-carbon multiple bonds constitutes
one of the most atom-economical means for the synthesis of cyclic amines and
imines. In the case of alkene, diene,
and allene hydroamination,
a new stereogenic center is formed during
carbon-nitrogen bond construction. This raises an important synthetic
opportunity for asymmetric catalysis leading to enantiomerically
enriched heterocycles.
During the proceeding grant
period, we have successfully synthesized and begun to evaluate two distinct
sets of optically active proligands (e.g., 1a-c
and 2) that will lead to complexes
in which the chelated metal is endowed with enhanced
electron density. Proligands 1a-c represent a conceptually new class of axially chiral bis(b-diiminate)s, whereas 2 is noteworthy by possessing external contact ligand chirality that will permit the evaluation of this
characteristic on enantioselectivity (Scheme 1).
Given the high reactivity exhibited
by the Zr(IV)¥NPS
chelates in the intramolecular
hydroamination of allenes,5 a series
of experiments are being performed
using precatalysts derived from Zr(NMe2)4
and the new chiral proligands
described above. A comparison of the ee's obtained
with these complexes to those of the corresponding Y, Lu and Sc chelates in the cyclization of aminoallenes 8a
and 8b should be mechanistically
interesting in that hydroaminations involving group 4
metals proceed via imido complexes6 as opposed to amido complexes,
as in the case of group 3 metals. In addition, a direct comparison of Sc(III) and Zr(IV) in
this context will be revealing as the covalent radii of these two metals are
similar. Irrespective of the viability of the Sc(III)
complexes generated from 1a-c and 2 as hydroamination catalysts, the use of
these proligands as their Y(III) complexes for
asymmetric hydroamination
will be revealing in terms of the electronic prerogatives of this metal vis a vis enantiocontrol. The evaluation of various group 3 and Zr(IV)
complexes of the above proligands for the asymmetric hydroamination of representative aminoalkenes
is currently underway.
The
professional impact of this research for me has been a far better appreciation
for, and consequently understanding of, those factors that influence the design
and synthesis of high-performance ligands for
asymmetric catalysis. My collaborator on this project has benefited
significantly from learning the specialized techniques of manipulating air and
moisture sensitive compounds in synthesis.
References
1(a). Nickel-Catalyzed
Transformations of 2,1-Benzisoxazoles with Organozinc
Reagents Baum, J. S.; Condon, M. E.; Shook, D. A. J. Org. Chem. 1987, 52, 2983. Bridgehead Nitrogen Heterocycles
which Contain the Quinazoline Moiety-Synthesis
and Cycloaddition of 1,2-Dihydroquinazoline 3-Oxides.
Org. Biomol.
Chem. 2005, 3, 4351. (b). Synthesis of Enantiomerically
Pure N-tert-Butane
Sulfinyl Imines (tert-Butanesulfinimines) by the
Direct Condensation of tert-Butanesulfinamide
with Aldehydes and Ketones.
Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman,
J. A. J. Org. Chem. 1999, 64, 1278. (c). Asymmetric Induction. 2.1 Enantioselective Alkylation of Cyclohexanone
via a Chiral Enamine. Whitesell, J.
K.; Felman, S. W. J.
Org. Chem. 1977, 42, 1663.
2. Enantioselective Intramolecular Alkene Hydroaminations Catalyzed by Yttrium Complexes of Axially Chiral Bis(thiolate) Ligands.
Kim, J. Y.; Livinghouse, T. Org. Lett. 2005, 7, 1737.
3(a). Direct 1H NMR Assay of
the Enantiomeric Composition of Amines and b-Amino Alcohols Using O-Acetyl Mandelic Acid as a Chiral
Solvating Agent. Parker, D.; Yaylor,
R. J. Tetrahedron 1987, 43, 5451. (b). Zirconium catalysed
enantioselective hydroamination/cyclisation.
Knight, P. D.; Munslow, I.; O'Shaughnessy, P. N.;
Scott, P. J. Chem. Soc. Chem. Commun. 2004,
894.
4. Highly Stereoselective Intramolecular Hydroamination/Cyclization of Conjugated Aminodienes Catalyzed by Organolanthanides.
Marks, T. J.; Hong, S. J.
Am. Chem. Soc. 2002, 124, 7866.
5. Intramolecular
Hydroaminations of Aminoalkynes
Catalysed by Yttrium Complexes and Aminoallenes Catalyzed by Zirconium complexes. Kim. H.;
Livinghouse, T.; Dong, S.; Lee, P. H. Bull. Korean
Chem. Soc. 2007, 28, 1127.
6. Development of the
Ti-Catalyzed Intramolecular Hydroamination
of Alkynes. Doye, S. Synlett. 2004, 1653 and
references therein.
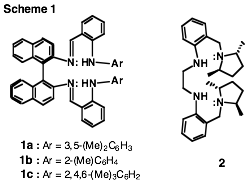
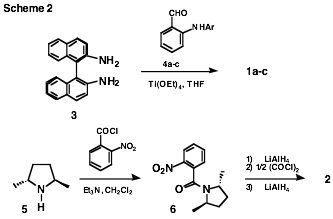 The above proligands
were readily accessible by the following routes. Condensation of [R- (or S-)]-1,1'-binaphthyl-2,2'-diamine (3) with 2 equiv. of the appropriate and
readily prepared 2-(arylamino)benzaldehyde
4a-c1a (Ti(OEt)4, THF)1b
provided 1a-c with high efficiency. Proligand 2 was derived from acylation
of (R,R-)-2,5-dimethylpyrrolidine
(5) (that is now readily available by our methodology, vide supra, followed
by resolution with mandelic acid1c)
with 2-nitrobenzoyl chloride to give 6.
Exhaustive reduction of 6 followed
by acylation of the resulting aniline with oxalyl chloride (0.5 equiv, i-Pr2NEt as an HCl scavenger)
and final reduction (LiAlH4) then delivered 2 (Scheme 2).
The above proligands
were readily accessible by the following routes. Condensation of [R- (or S-)]-1,1'-binaphthyl-2,2'-diamine (3) with 2 equiv. of the appropriate and
readily prepared 2-(arylamino)benzaldehyde
4a-c1a (Ti(OEt)4, THF)1b
provided 1a-c with high efficiency. Proligand 2 was derived from acylation
of (R,R-)-2,5-dimethylpyrrolidine
(5) (that is now readily available by our methodology, vide supra, followed
by resolution with mandelic acid1c)
with 2-nitrobenzoyl chloride to give 6.
Exhaustive reduction of 6 followed
by acylation of the resulting aniline with oxalyl chloride (0.5 equiv, i-Pr2NEt as an HCl scavenger)
and final reduction (LiAlH4) then delivered 2 (Scheme 2).
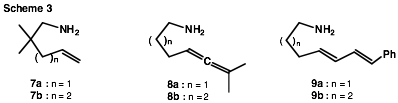 Asymmetric intramolecular
hydroaminations involving the aminoalkenes
7a,b, the aminoallenes
8a,b, and the aminodienes 9a,b are currently being carried
out as previously described2 and the resulting ee's
are being determined by 1H
NMR after diastereomeric salt formation using optically active
O-acetylmandelic acid3a or as a Mosher's
amide3b (Scheme 3). The selection of the E,E-dienes 9a,b
with terminal phenyl substitution is based on the known4
predisposition of this substrate type to undergo 1,2- (and not 1,4-) N-H
addition to the conjugated p-system.
Asymmetric intramolecular
hydroaminations involving the aminoalkenes
7a,b, the aminoallenes
8a,b, and the aminodienes 9a,b are currently being carried
out as previously described2 and the resulting ee's
are being determined by 1H
NMR after diastereomeric salt formation using optically active
O-acetylmandelic acid3a or as a Mosher's
amide3b (Scheme 3). The selection of the E,E-dienes 9a,b
with terminal phenyl substitution is based on the known4
predisposition of this substrate type to undergo 1,2- (and not 1,4-) N-H
addition to the conjugated p-system.



