Reports: GB4
46907-GB4 Development of Peptide Based Biocatalysts for the Desulfurization of Dibenzothiophene
The presence of sulfur in hydrocarbon fuels has been a focal point of recent nation and international legislation largely due to environmental and health concerns. The combustion of sulfur generates sulfur oxyacids that contribute to deforestation and cardiopulmonary disease. The current technology for reducing sulfur content in fuels, hydrodesulfurization, cannot eliminate recalcitrant aromatic sulfur compounds. Discovery of bacteria that metabolize aromatic sulfur heterocycles has led to significant interest in biocatalytic desulfurization. However, meeting the aqueous environment needed by the organism, while effectively removing sulfur from fuel stocks has not been achieved.
| Figure 1. Metabolic pathway for the desulfurization of DBT
|
 In
the biological metabolism of DBT uses DszB, a desulfurase, and is though to
involve a number of flavodoxins, or related enzymes. The crystal structure of
DszB, from Rhodococcus erythropolis,
was recently solved, revealing that binding occurs from contact with six
primary residues, three of which display an edge-face aromatic interaction. Flavodoxin
binding to flavin commonly involves aromatic residues as well. Favorable
electrostatic interactions arise from the proximity of the electron rich
centroid of the ring and the electron poor hydrogens. The ubiquitous presence
of aromatic interactions in enzymatic action on DBT has biased design of a
molecular receptor.
In
the biological metabolism of DBT uses DszB, a desulfurase, and is though to
involve a number of flavodoxins, or related enzymes. The crystal structure of
DszB, from Rhodococcus erythropolis,
was recently solved, revealing that binding occurs from contact with six
primary residues, three of which display an edge-face aromatic interaction. Flavodoxin
binding to flavin commonly involves aromatic residues as well. Favorable
electrostatic interactions arise from the proximity of the electron rich
centroid of the ring and the electron poor hydrogens. The ubiquitous presence
of aromatic interactions in enzymatic action on DBT has biased design of a
molecular receptor.
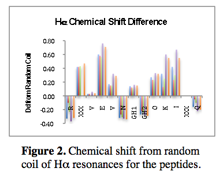
The peptide biocatalyst includes a number of essential features: a regular structure, modifiable sidechains and aromatic residues to interaction with DBT. Progress towards peptide receptor exploration has resulted in four possible sequence designs, cyclic and acyclic versions of diagonal and lateral sidechains. Initially, in order to guarantee structural rigidity all peptides were macrocyclized with a disulfide bond from terminal cystine residues. However, many peptides demonstrated sufficient stability to consider eliminating the disulfide bond to allow for greater positional freedom. Figure 2 shows the shift from random coil of the Ha resonances of a representative peptide. This peptide is 77% folded and displays significant Ha alteration (more than 0.1 ppm shift is considered an indication of structure). Disulfide bonds are still used in the event that a peptide is not sufficiently folded. In this manner greater conformational flexibility is available to maximize interactions with a substrate.
Figure 3. The structure of TrpZip a designed, 12 amino acid b-hairpin, with diagonal tryptophan residues as an aromatic cleft. The b-hairpin backbone is shown in green and the Trp sidechains as space filling spheres.
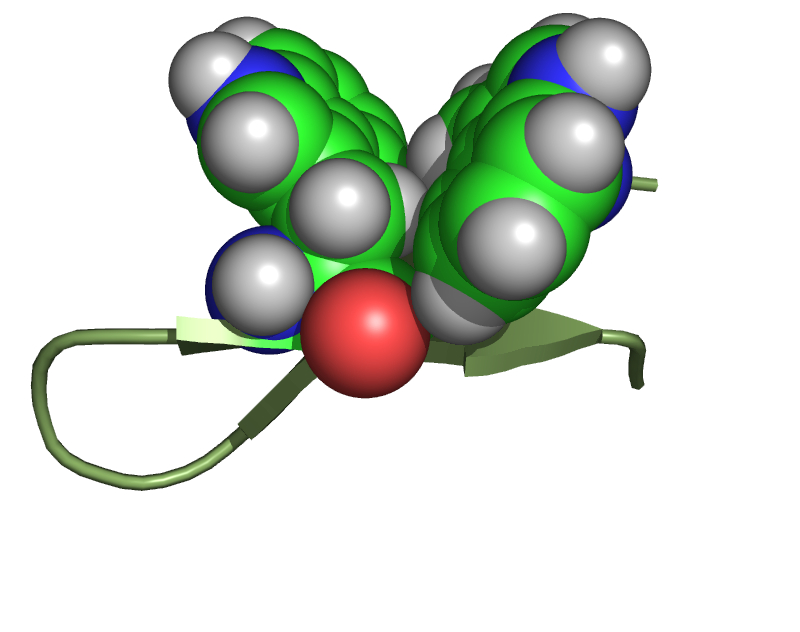
The positioning of the aromatic sidechains has been varied to determine the impact on binding site. Initially the aromatic residues were positioned diagonally (i, j-2) within the peptide (Figure 3). A number of peptides were synthesized and characterized utilizing this structural motif (RFVKVNGOFIKQ, RWVTVNGOWITQ, RWVKVNGOWIKQ, and RWVKVNGOWIFQ). All of these peptides were well folded (greater than 60%) and have been characterized. A number of technical problems have been found from early investigations into the binding potential of these peptides.
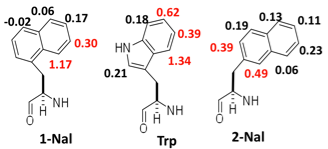
Figure 4. The shift from random coil of the indicated aromatic resonances. Red indicates maximum shift. Peptide sequence is RXVEVNGOKIFQ.
 The
excitation band for Trp is altered within the diagonal geometry such that it
overlaps with the DBT absorbance band. As such binding data cannot be obtained
from fluorescence spectroscopy. Analysis with NMR based binding studies in much
slower and ongoing. The phenylalanine containing peptides are expected to bind
the weakest, but could be amenable to fluorescence analysis and will be
investigated. The aromatic rings are not sufficiently close to interaction with
one another, but can bind to an intercalated substrate in a sandwich motif.
While this offers an efficient method for packing it appears to confer only
offset stacked geometries.
The
excitation band for Trp is altered within the diagonal geometry such that it
overlaps with the DBT absorbance band. As such binding data cannot be obtained
from fluorescence spectroscopy. Analysis with NMR based binding studies in much
slower and ongoing. The phenylalanine containing peptides are expected to bind
the weakest, but could be amenable to fluorescence analysis and will be
investigated. The aromatic rings are not sufficiently close to interaction with
one another, but can bind to an intercalated substrate in a sandwich motif.
While this offers an efficient method for packing it appears to confer only
offset stacked geometries.
To diversify the accessible geometries for interacting with substrates a variation is being explored. Laterally disposed aromatic residues (i, j) are sufficiently close to interact with each other, which seems to preclude them
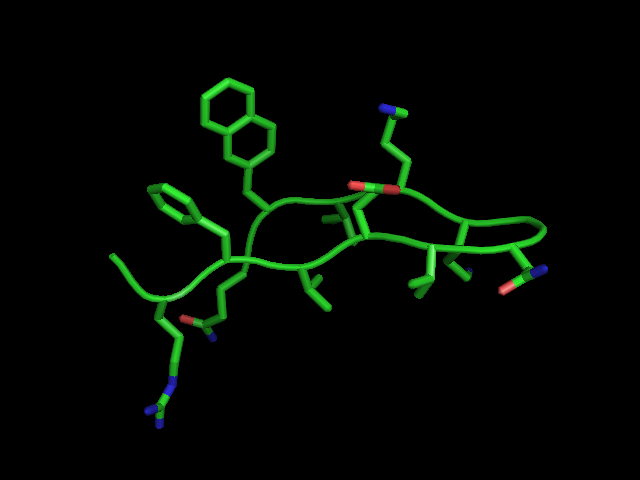
Figure 5. Preliminary structure of laterally interacting aromatic residues.
 from
subsequent substrate interactions. However, our studies indicate that the
lateral interaction occurs predominantly through one or two hydrogens via an
edge-face geometry. Figure 4 shows the shift of
specific aromatic resonances from random coil when laterally positioned with a
phenylalanine. What is interesting from a design perspective is that
1-naphthylalnine and tryptophan dive very similar shifts. This suggests that
these two rings adopt similar geometries. A subtle shift in the point of attachment
to 2-naphthylalanine gives a very different set of shifts. For both 1- and
2-naphthylalanine the majority of the aromatic ring is not involved in a
lateral aromatic interaction. A simmulated annealing reveals that the aromatic
rings are edge-face with a cleft for edge-face type interactions with a
substrate (Figure 5).
from
subsequent substrate interactions. However, our studies indicate that the
lateral interaction occurs predominantly through one or two hydrogens via an
edge-face geometry. Figure 4 shows the shift of
specific aromatic resonances from random coil when laterally positioned with a
phenylalanine. What is interesting from a design perspective is that
1-naphthylalnine and tryptophan dive very similar shifts. This suggests that
these two rings adopt similar geometries. A subtle shift in the point of attachment
to 2-naphthylalanine gives a very different set of shifts. For both 1- and
2-naphthylalanine the majority of the aromatic ring is not involved in a
lateral aromatic interaction. A simmulated annealing reveals that the aromatic
rings are edge-face with a cleft for edge-face type interactions with a
substrate (Figure 5).
Future direction: Analysis of the binding of peptide to DBT and DBTO2 with the diagonal aromatic residues is ongoing. A computational structure will be generated for the lateral polycyclic aromatic peptides from NOE restraints. Binding data will then be generated with the lateral aromatic peptides. After this initial round of diagonal and lateral structural and binding analysis an investigation into the redox potential of DBT and perturbations from binding will begin.



